
The del(5q) cytogenetic abnormality was first described by Van den Berghe et al. in 1974.1 The original manuscript described five patients with a macrocytic anemia, dyserythropoiesis with erythroid hypoplasia, normal to elevated platelet count, hypolobulated megakaryocytes and an interstitial deletion involving the long arm of chromosome 5. However, a deletion of chromosome 5q in myelodysplastic syndrome (MDS) does not necessarily equate to the clinically described 5q- syndrome because, by definition, the 5q- syndrome is a distinct entity with the aforementioned clinical characteristics.2 As we routinely evaluate patients with suspicion of MDS, some unusual clinical situations have emerged in our clinical practice.
In December 2013, a 65-year-old asymptomatic woman was referred to us to investigate isolated leukopenia. The physical exam was normal. The complete blood count showed mild leukopenia (white blood cells 2.65×109/L – neutrophils 36%, eosinophils 8%, basophils 0%, lymphocytes 48% and monocytes 8%), hemoglobin 12.3g/dL (normal range 11.5–16.4g/dL), mean corpuscular volume (MCV) 101.1fL and platelets 326×109/L. The patient was submitted to bone marrow (BM) aspiration and biopsy, as well as cytogenetic analysis by G-banding.
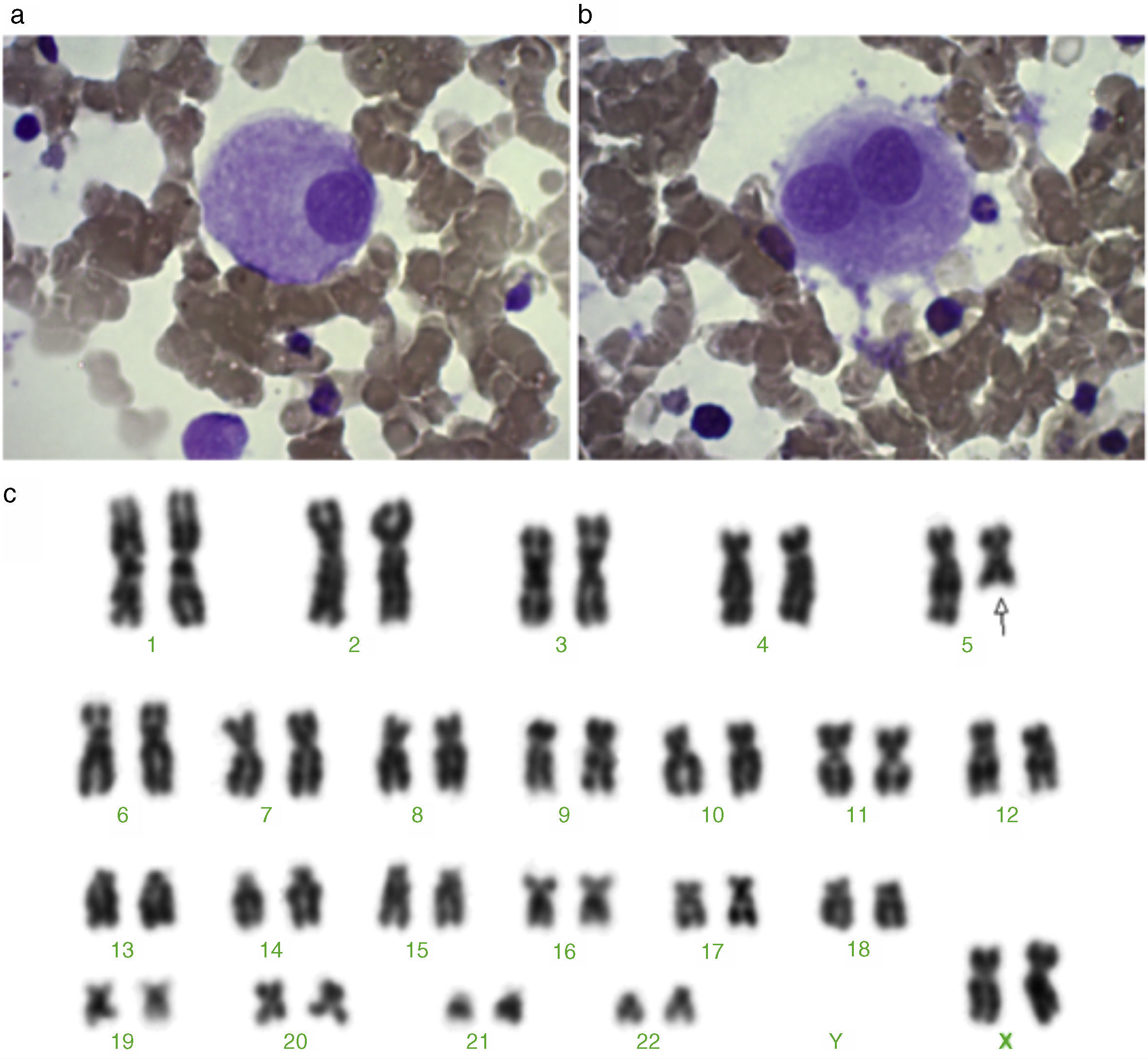
The BM smear revealed a normocellular erythrocyte lineage, characterized by the presence of dysplasia in 12% of cells. The megakaryocytic lineage was hypercellular and characterized by dysplasia in 28% of cells. The morphologically abnormal megakaryocytes exhibited a non-lobulated, eccentric nucleus (Figure 1a). Occasionally, ‘dumbbell-type’ megakaryocytes were identified (Figure 1b).
 Megakaryocyte with a non-lobulated nucleus. (b) A ‘dumbbell-type’ megakaryocyte. (c) Karyotype showing a large terminal deletion of chromosome 5 – 46,XX,del(5)(q22).")
The BM biopsy showed overall hypocellularity and confirmed the significant dysmegakaryopoiesis. Dyserythropoiesis was characterized by megaloblastoid erythroblasts (<10%). The granulocytic series showed no significant abnormalities.
Following the recommendations of the International System for Human Cytogenetic Nomenclature (ISCN 2013), the cytogenetic analysis demonstrated a karyotype with a large interstitial deletion in the long arm of chromosome 5 (5q22) in 50% of 16 analyzed metaphases (46,XX,del(5)(q22)[8]/46,XX[8]) (Figure 1c).
The patient's last medical consultation took place in June 2015 and she was asymptomatic. She continues to be followed by her local hematologist.
In March 2015, the hemoglobin level dropped to 10.6g/dL. The last complete blood count dated June 2015 showed white blood cells of 1.64×109/L – neutrophils 23.9%, eosinophils 5.2%, basophils 0.5%, lymphocytes 61.6%, monocytes 8.8% – hemoglobin 9.9g/dL, MCV 109.1fL, and platelets 195×109/L. The median value and the minimum–maximum value for hemoglobin, and neutrophil and platelet counts during the period of the follow up were 11.5g/dL (9.9–12g/dL), 1.003×109/L (0.391–1.569×109/L) and 239×109/L (184–281×109/L), respectively.
Myelodysplastic syndromes are a group of clonal hematopoietic disorders with diverse clinical presentations.3 A subset of patients have an isolated deletion of chromosome 5q, known as 5q- syndrome, which is characterized by a distinct set of features including severe macrocytic anemia, normal or slightly decreased neutrophil count, normal or elevated platelet count and, in bone marrow, the presence of hypolobulated megakaryocytes.2
Our patient has a large 5q deletion (5q22), which includes both commonly deleted regions, 5q31 and 5q32-33. One of the genes lost by these deletions is the adenomatous polyposis coli (APC) gene that may contribute to the pathogenesis of del(5q) MDS due to increased beta-catenin activity.4 Another important affected gene is the Ribosomal Protein S14 (RPS14) gene, located in the 5q31-q33 locus. RPS14 haploinsufficiency is associated with increased apoptosis of erythroid precursors and ineffective erythropoiesis. However, contrary to expectations, our patient had no anemia at diagnosis and, thus, we suggest that additional molecular abnormalities arising over time, such as somatic mutations in individual genes and epigenetic alterations, may also contribute to the many different clinical phenotypes of patients with del(5q). Each of these abnormalities has the potential to alter blood counts, the percentage of blast cells in the BM and overall survival.4
Our patient presented with some BM abnormalities typically found in patients with the classical 5q- syndrome as, for example, the almost pathognomonic non-lobulated and ‘dumbbell-type’ megakaryocytes. On the other hand, severe transfusion-dependent macrocytic anemia, which is a characteristic mark of this syndrome, is absent. Thus, given the clinical and laboratory findings of this case, it was not possible to unequivocally define between the diagnosis of classical 5q- syndrome as described by Van den Berghe1 and MDS with isolated del(5q).
However, we concluded that, at present, this differential diagnosis is not necessary in the clinical practice. This is because, as demonstrated by recent evidence,5 therapy with the orally active thalidomide analog, lenalidomide, can be used to treat patients with both conditions as it is indicated for subjects with del(5q) and symptomatic anemia.
Finally, the effectiveness of lenalidomide in classical 5q- syndrome and MDS with isolated del(5q) supports the findings in the World Health Organization (WHO) 2008 classification, that does not require specific differential diagnosis between the entities. Nevertheless, it is still possible that each disease may have different approaches in the future, as new molecular methods have contributed to better define the topographic features of the genomic lesions involved in del(5q) over recent years.
Conflicts of interestThe authors declare no conflicts of interest.
ConsentInformed consent was obtained from the patient.


