

Even though hemolytic anemias (HAs) are not very common, their diagnosis remains a big challenge for hematologists and clinicians. We hope that this summary will contribute with valuable information about a subject that has been little described in the medical literature, and will help to clarify the diagnostic approach to guide specific treatment depending on the causative condition.
It is known that HAs are a group of disorders characterized by a premature red blood cell (RBC) destruction (less than 120 days),1,2 that exceeds the compensatory capacity of the bone marrow to increase RBC production and keep up with the loss.1–3 Usually HAs are diagnosed through laboratory tests, however, the patient's history and physical examination are crucial as they provide important information about the presence of hemolysis and its probable etiology.3,4 For example, if in addition to the classic symptoms of anemia (paleness, fatigue, dyspnea, palpitations), findings such as familial or personal history of jaundice, exposure to toxics, leg ulcers, lymphadenopathies, hepatomegaly or splenomegaly may help to elucidate the cause of the anemia.1,3,4
Regarding the etiology, HAs can be classified as inherited or acquired and when considering the site of hemolysis, RBCs can be destroyed in the circulation (intravascular) or within macrophages in the spleen or liver (extravascular). From the clinical perspective, HAs can be acute or chronic and according to the location of the abnormality responsible for the hemolysis, they may be due to intrinsic (intracorpuscular) or extrinsic (extracorpuscular) defects.2–4 It is important to mention that most intrinsic defects are inherited, and most extrinsic ones are acquired,2 however, there are some exceptions to this rule. For example, paroxysmal nocturnal hemoglobinuria (PNH) is an acquired HA produced by an intrinsic defect4 and glucose-6-phosphate dehydrogenase (G6PD) deficiency is an inherited intrinsic defect that is triggered by an external factor.2
Although there are different ways of approaching the diagnosis of HAs, it is first necessary to identify the patient with HA and collect data on hemolysis. The destruction of RBCs in HAs is characterized by an increased breakdown of hemoglobin which results in unconjugated hyperbilirubinemia clinically evidenced by jaundice, increased lactate dehydrogenase (cellular destruction), and reticulocytosis, which is a normal compensatory response of the bone marrow to the RBC loss. Additionally, decreased levels of plasma haptoglobin, a marker of RBC destruction, are evidenced1–3 regardless the site of hemolysis (intravascular or extravascular).5 In cases of severe acute intravascular HAs, the haptoglobin-binding capacity reaches its saturation point, and free hemoglobin is filtered by the glomerulus and hemoglobinuria is seen.3,6 Also, hemosiderinuria may be present in long-term intravascular hemolysis.3,6 On the other hand, if the hemolysis is extravascular, urobilinogen may appear in the urine or feces.6
Once we have a presumptive diagnosis, a multi-step procedure is required,4 beginning with a direct antiglobulin test (DAT) or direct Coombs test.6,7 This is the exam of choice because it will classify the hemolysis into an immune or non-immune etiology.6,7 Although DAT helps to categorize HAs into two groups, results must be interpreted in the context of the clinical conditions as some conditions may be incorrectly classified.7,8 This is why the DAT should be followed by a peripheral blood smear (PBS) and an investigation of the family history.6 A simple approach to identify and classify HAs based on the works of González Mesones et al.6 and others3,4,8 is presented in Figure 1.

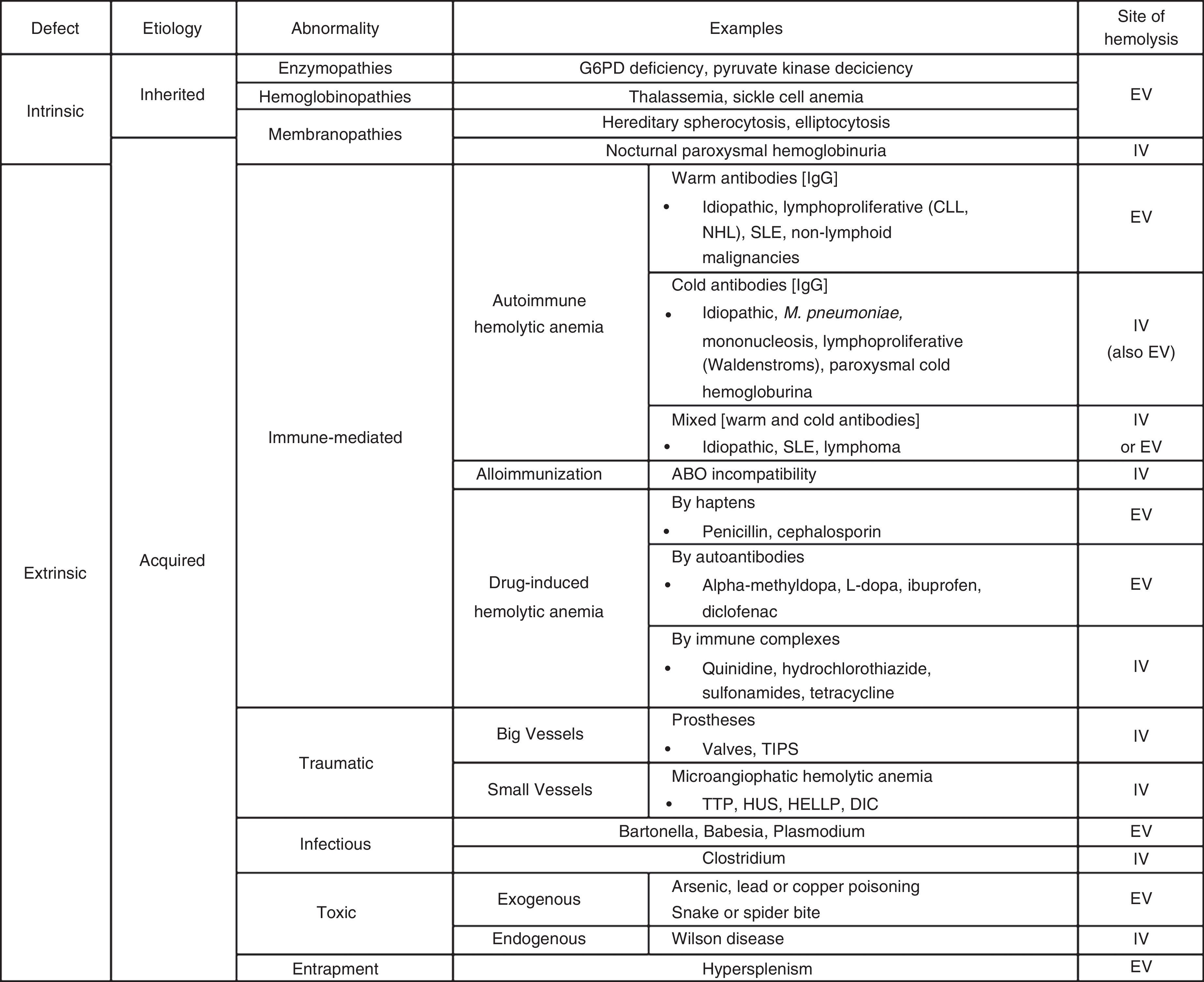
In cases of immune HAs (DAT positive), the diagnostic possibilities are limited to the presence of autoantibodies (warm, cold or mixed), alloimmunization (as in transfusion incompatibility), or drug-related (hemolysis by haptens, autoantibodies or immune complexes). Reviewing the PBS is a critical step in the evaluation of non-immune HAs (DAT negative). Certain morphological abnormalities of RBCs correlate with different pathologies. For example, schistocytes are related to thrombotic microangiopathies or cardiac prosthetic valves, spherocytes with hereditary spherocytosis, sickled cells with sickle cell disease, elliptocytes with hereditary elliptocytosis, echinocytes with pyruvate kinase deficiency, Heinz bodies with G6PD deficiency, and basophilic stipplings with lead poisoning, thalassemia, and Wilson's disease, among others.4 Thus, if the PBS is abnormal, the family history may identify a congenital HA including disorders of enzymes (G6PD deficiency, pyruvate kinase deficiency) hemoglobin (thalassemia, sickle cell anemia) or of the membrane (hereditary spherocytosis, elliptocytosis). Likewise, traumatic hemolysis (of big or small vessels), infectious (Bartonella, Babesia, Plasmodium) or toxic (exogenous or endogenous) pathologies should be taken into account if there is an abnormal PBS in the absence of family history. In contrast, if the PBS is normal, hypersplenism or PNH should be considered. A detailed list of the most common causes of hemolysis classified by type is presented in Table 1.
The most common causes of hemolysis classified by type.
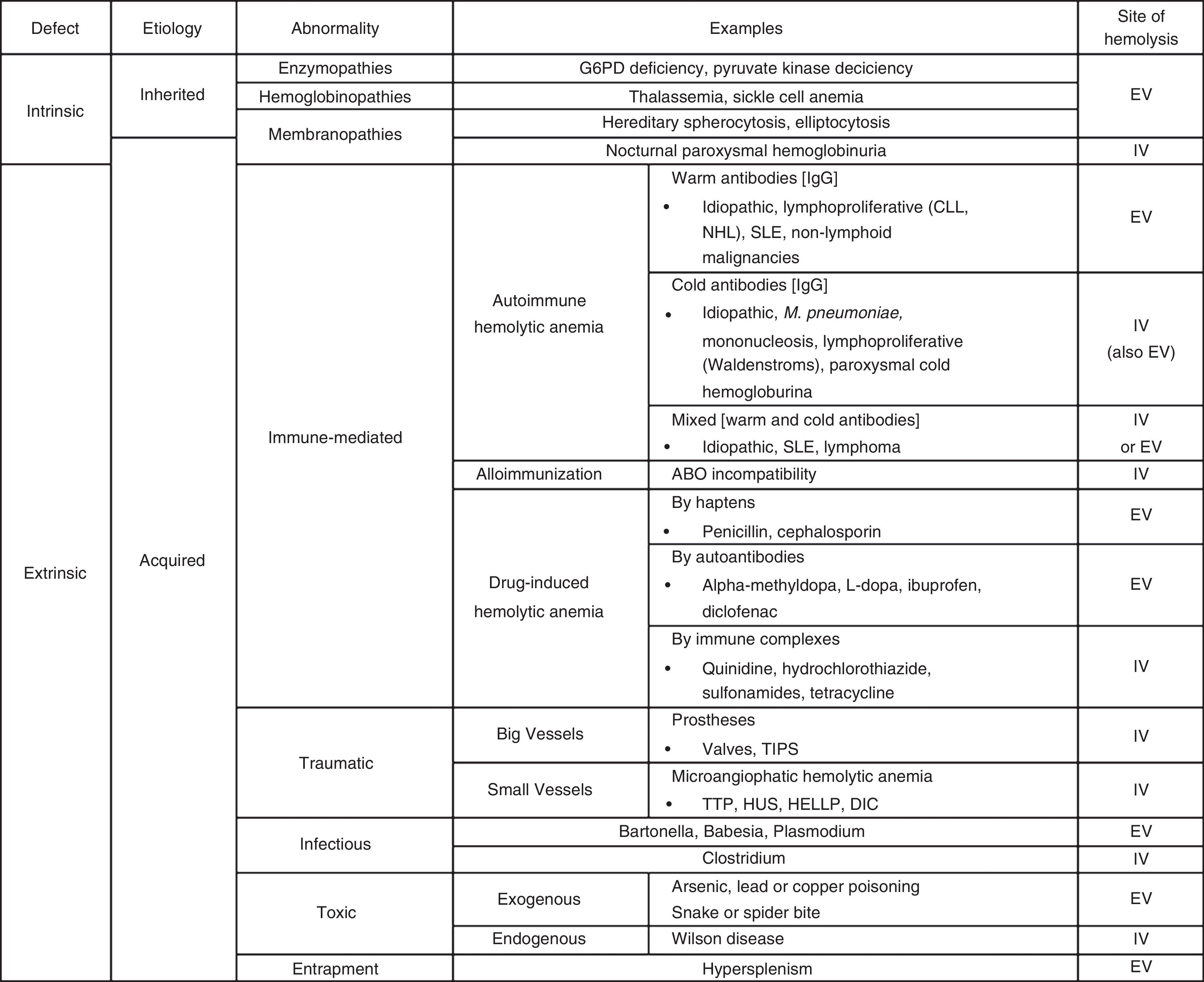
G6PD: glucose-6-phosphate dehydrogenase; CLL: chronic lymphocytic leukemia; NHL: non-Hodgkin lymphoma; SLE: systemic lupus erythematous; TIPS: transjugular intrahepatic portosystemic shunt; TTP: thrombotic thrombocytopenic purpura; HUS: hemolytic uremic syndrome; HELLP: hemolysis, elevated liver enzyme levels, and low platelet levels; DIC: disseminated intravascular coagulation; EV: extravascular: IV: intravascular.
The authors declare no conflicts of interest.



