

Hemoglobinopathy Sβ-thalassemia (HbSβ-thal) has a wide range of clinical and laboratory severity. There is limited information on the natural history of HbSβ-thal and its modulating factors. We described the molecular, hematological, and clinical characteristics of a cohort of children with HbSβ-thal and estimated its incidence in Minas Gerais, Brazil.
MethodsLaboratory and clinical data were retrieved from medical records. Molecular analysis was performed by HBB gene sequencing, PCR-RFLP, gap-PCR, and MLPA.
ResultsEighty-nine children were included in the study. Fourteen alleles of β-thal mutations were identified. The incidence of HbSβ-thal in the state was 1 per 22,250 newborns. The most common βS-haplotypes were CAR and Benin. The most frequent βthal-haplotypes were V, II, and I. Coexistence of 3.7 kb HBA1/HBA2 deletion was present in 21.3 % of children. β-thalassemia mutations were associated with several clinical and laboratory features. In general, the incidence of clinical events per 100 patient-years was similar for children with HbSβ0-thal, IVS-I-5 G>A, and IVS-I-110 G>A. Children with HbSβ+-intermediate phenotypes had a more severe laboratory and clinical profile when compared with those with HbSβ+-mild ones. βS-haplotypes and α-thalassemia did not meaningfully influence the phenotype of children with HbSβ-thal.
ConclusionThe early identification of β-thalassemia alleles may help the clinical management of these children.
Sickle cell disease (SCD) is a hereditary disorder that significantly affects the world population. In Brazil, it is considered a public health issue. Although primarily determined by a single mutation in the beta globin gene [HBB:c.20A>T (p.Glu6Val)], SCD has a wide phenotypic variation attributed to genetic, environmental modulators and interactions with other variant hemoglobins (Hb).1 The most frequent subtypes of SCD in Brazil are Hb SS, Hb SC, and Hb Sβ-thalassemia (HbSβ-thal).2
HbSβ-thal is a subtype of SCD characterized by clinical and laboratory heterogeneity, mainly due to different β-thal mutations and, consequently, null or different levels of residual production of HbA. HbSβ-thal is classified according to the gene expression of the β-thal allele into HbSβ+-thal (residual HbA) or HbSβ0-thal (absence of HbA).3-5 In general, HbSβ+-thal is considered to be a milder or intermediate phenotypical subtype of SCD while HbSβ0-thal is similar to sickle cell anemia (SCA).
Although the β-thal mutation can partly explain the phenotypic variation, there is still a wide variability in the clinical severity of HbSβ-thal.3,6 Co-inherence of alpha-thalassemia (α-thal) and beta-globin gene cluster haplotypes may influence the severity of this subtype of SCD. Haplotypes are well studied in many reports, mainly in SS patients.7 In previous papers, we demonstrated that co-inheritance of α-thal significantly decreased the risk of cerebrovascular disease in SS children and of acute splenic sequestration in SC children.8,9 However, the association of haplotypes or α-thal with the clinical course of HbSβ-thal has been only occasionaly reported.10
It is estimated that there are 60,000 people with SCD in Brazil, with a wide incidence variation between regions and states.2 However, the incidence of HbSβ-thal is unknown. The identification of disease burden and modulating factors in children with HbSβ-thal may help to provide a targeted clinical approach, resulting in a better quality of life and life expectancy. In this study, the natural history of HbSβ-thal in children from Minas Gerais, a southeastern state of Brazil, is described. Additionally, we estimated the incidence of HbSβ-thal in this state.
Material and methodsStudy design and settingThis was a retrospective cohort study involving children with HbSβ-thal diagnosed by the Newborn Screening Program (NSP) from Minas Gerais, Brazil. For the description of natural history, the sample consisted of children born between January 1999 and December 2015, screened by NSP and followed-up at the outpatient clinic of Hemominas, Blood Center of Minas Gerais. To study the incidence of HbSβ0-thal and HbSβ+-thal in Minas Gerais, all children screened by NSP in the state between January 2011 and December 2015 with a suggestive isoelectric (IEF) and HPLC profile of HbSβ-thal were included. In these cases, HBB sequencing was always done to confirm the genotype.
Eligibility criteriaInclusion criteria for participants with Hb Sβ-thal were: 1) children born between January 1999 and December 2015 screened by NSP with HbSβ-thal and followed up at the Blood Center; 2) Written informed consent obtained from participants’ parents or guardians and from children's assent when appropriate. Inclusion criteria to estimate the incidence of HbSβ-thal in the state were: all children born between January 2011 and December 2015 (five years) screened by NSP with HbSβ-thal, according to the previous described definition. This period limitation was necessary because allele-specific PCR reaction for codon 7 of HBB (GAG, GTG, or AAG) was only introduced as a NSP routine in March 2010. All babies with Hb FS or FSA detected by IEF and HPLC had, then, their HBB gene sequenced.
MeasurementsData collection instrumentsA medical record data abstraction was performed for all consenting participants. Medical records were reviewed by hematologists or research nurses under the supervision of hematologists. Clinical, laboratory, and treatment data were abstracted using standardized definitions of the clinical outcomes of SCD11 and entered into a specific database. The period of clinical follow-up of the study population was between 01/01/1999 (beginning of the cohort follow-up) and 01/01/2019 (end of the cohort follow-up). All children had at least 2.8 years at last clinical visit. Only clinical outcomes recorded before initiation of modifying therapies (hydroxyurea or chronic transfusion program) were considered.
Transcranial Doppler (TCD) examinations were all made and interpreted by a single expert. The Stroke Prevention Trial in the Sickle Cell Anemia study protocol12 was followed, with pulse TCD and a 2 MHz probe for a full Doppler test (EME TC 2000; Nicolet, Madison, WI, USA).
Molecular analysisGenomic DNA extraction from blood samples was performed using a commercial kit (QIAamp, DNA Blood Mini Kit, Qiagen, Hilden, Germany). The identification of β-Thal mutations was conducted by Sanger sequencing (Supplementary Methods).
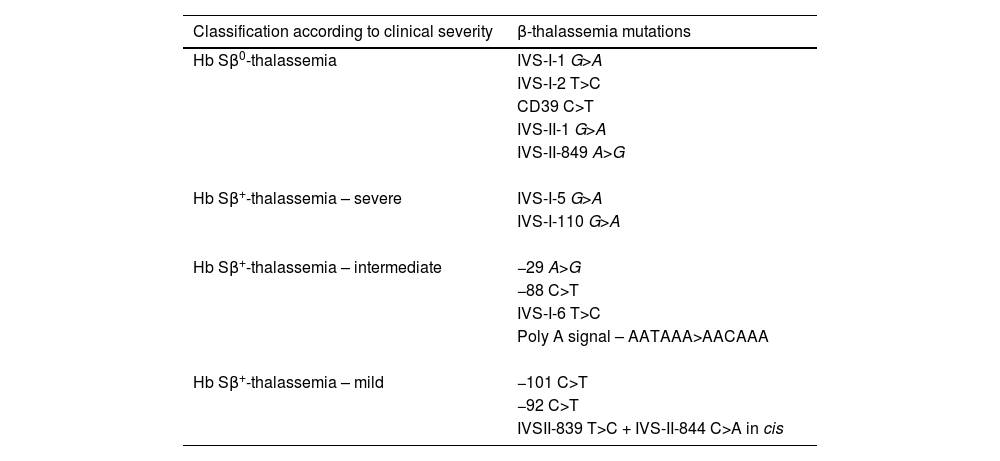
Children were classified as having HbSβ0-thal, severe HbSβ+-thal, intermediate HbSβ+-thal, and mild HbSβ+-thal, according to β-thalassemia mutation (Table 1). The classification was based on genotype-phenotype correlations that have been shown in this and previously published studies,3,4,13-22 as well as review of Hb variant databases.23,24
Classification of β-thalassemia mutations according to the clinical severity of the hemoglobinopathy Sβ-thalassemia in children.
βS and βThal haplotypes were determined by PCR with specific primers, followed by Restriction Fragment Length Polymorphism (RFLP) or Sanger sequencing. The primer sequences, PCR protocols, and RFLP conditions are shown in the Supplementary Table 1. Detection of HBA more frequent deletions (−α3.7, −α4.2, −−SEA, −−FIL, −−MED, −(α)20.5, and –THAI) and HBA triplication were carried out by multiplex gap PCR as described previously25; the primers annealing temperature was increased to 62 °C. Identification of the Corfu deletion in participants with IVSI-5 G>A was performed by Multiplex Ligation-dependent Probe Amplification.
Laboratory dataTotal Hb, mean corpuscular volume (MCV), mean corpuscular hemoglobin (MCH), white blood cell (WBC) count, reticulocyte, and platelet count were determined using electronic cell counter (CELL-DYN Ruby, Abbott Laboratories, Santa Clara, CA, USA). Hemoglobin fractions were quantified by high-performance liquid chromatography (Beta-thalassaemia Short Program on the Variant II analyzer, BioRad, Hercules, CA, USA). The mean of Hb, MCV, MCH, WBC, platelet, and reticulocyte counts were calculated for children above two years of age, and steady-state values were registered. Values obtained from tests performed after blood transfusion (up to 90 days) and during serious clinical illness (vaso-occlusive pain crisis, severe infection, acute splenic sequestration, and aplastic crisis) were disregarded. For children who underwent modifying therapies (hydroxyurea and/or chronic blood transfusion program), steady state values were determined from the data registered before initiating the therapy.
Statistical analysisContinuous variables were expressed as mean and standard error of the mean (SEM) or median and interquartile range (IQ) as appropriate. Categorical variables were expressed as percentages of total and 95 % confidence interval (CI). Normal distribution of continuous variables was checked by the Kolmogorov-Smirnov test. The univariate analysis of continuous variables was performed using the unpaired t-test, except for those variables with deviation from normal distribution, in which case Mann-Whitney U test was used. Univariate associations between categorical variables were evaluated using Fisher's exact test. The incidence of clinical outcomes was reported by relative rates to 100 patient-years (pt-yrs) and 95 % CI. The incidence rate ratio (IRR) of clinical outcomes between groups was compared using OpenEpi online software,26 by Fisher's Exact Test. Cumulative risk of acute splenic sequestration was estimated by using a Kaplan-Meier method [function (1− survival)] and the log rank test was used to compare different subgroups based on risk factors. Birth date determined entry into the study program. Death by any causes, and last clinical visit without acute splenic sequestration until January 2019 (end of follow-up) were reasons for censored observations. Tests with probability p < 0.05 were considered significant. Statistical analyses were performed with the SPSS 20.0 (SPSS Inc.; Chicago, IL, USA).
ResultsHb Sβ-thal incidence in the state of Minas GeraisFrom 1,179,389 neonates born between 01-Jan-2011 and 31-Dec-2015, 53 were diagnosed with HbSβ-thal. Out of these 53 babies, 26 (49.1 %) had HbSβ0-thal and 27 (50.9 %) HbSβ+-Thal (all subtypes). The overall incidence of HbSβ-thal in the state was 1:22,250 (95 % CI, 1:17,530 to 1:30,450). The incidence of HbSβ0-thal was 1:45,360 (95 % CI, 1:32,770 to 1:73,680) and that of HbSβ+-thal was 1:43,680 (95 % CI, 1:31,720 to 1:70,140).
Cohort characteristicsFrom January 1999 to December 2015, 127 neonates screened with a suggestive clinical and laboratory picture of HbSβ-thal were sent to clinical follow-up at the Blood Center. Out of these 127 children, two individuals did not consent to participating in this study, one was deceased, eight were transferred to other outpatient clinics, and 27 had genotypes other than HbSβ-thal (HbSS, HbS/HPFH, HbS/Porto Alegre, HbS/Saki, HbS/Köln, HbS/Yaizu, and sickle cell trait). Therefore, 89 children with HbSβ-thal were included in the present study.
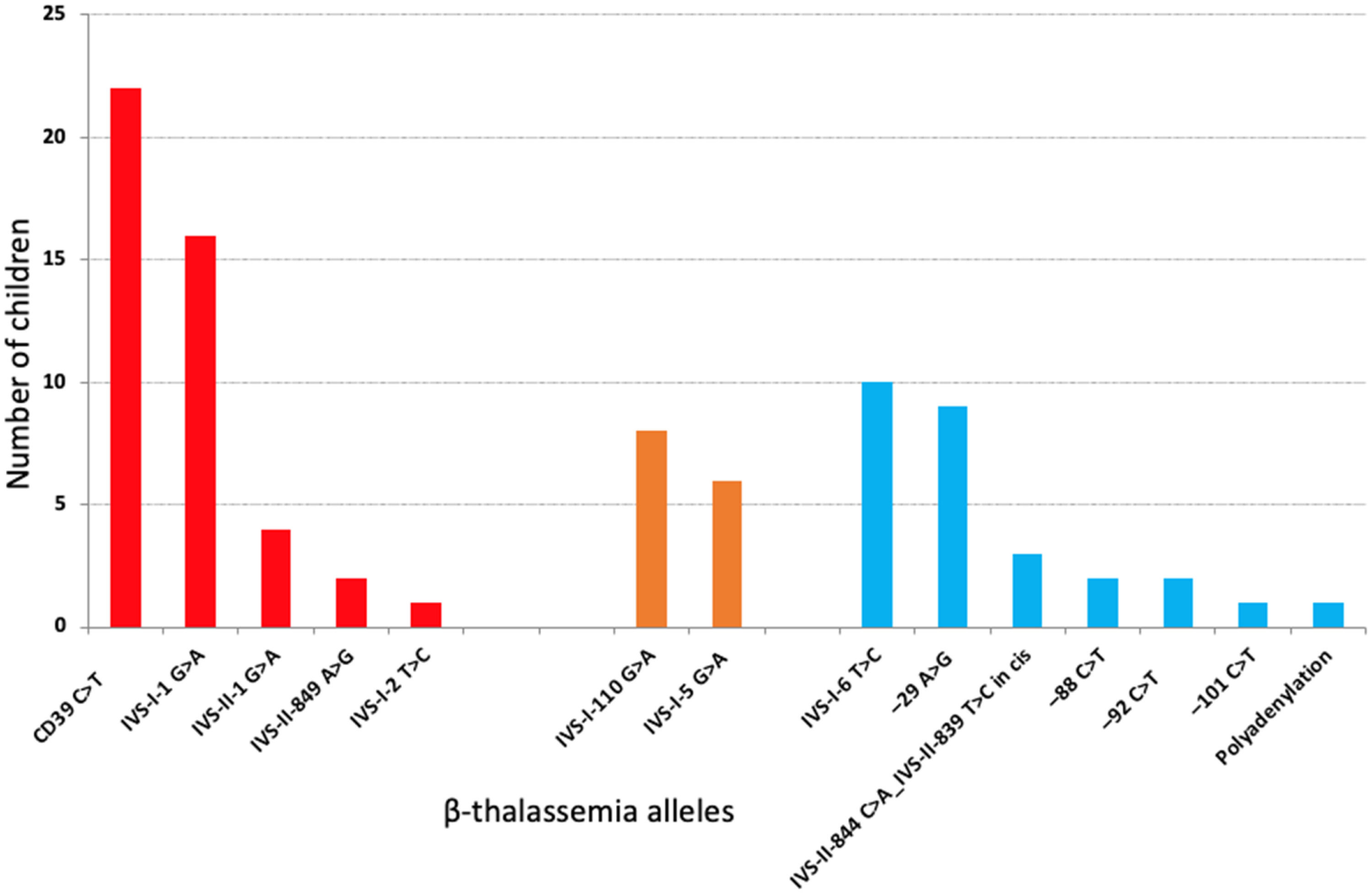
The median age at last clinical visit was 11.7 y (IQ range, 8.3 y); 52.8 % were females. The median follow-up up to starting HU or CBT was 9.8 years (IQ range: 9.0 years), providing 805.8 patient-years. Twenty-nine (32.6 %) children received hydroxyurea (HU) therapy and three (3.4 %), blood transfusion (CBT) and HU during follow-up. There were 14 different types of β-thal causative variants (Figure 1). The Corfu delta-beta thalassemia deletion was not detected in any of the five families with IVS-I-5 G>A mutation (NG_000007.3:g.57237_64443del7207).

Frequency of 14 different types of β-thal causative variants in 87 children with hemoglobinopathy S/beta-thalassemia. In two children, sequencing did not disclose any mutation. In red, S/beta0-thalassemia; in orange, severe S/beta+-thalassemia; in blue, intermediate or mild S/beta+-thalassemia.
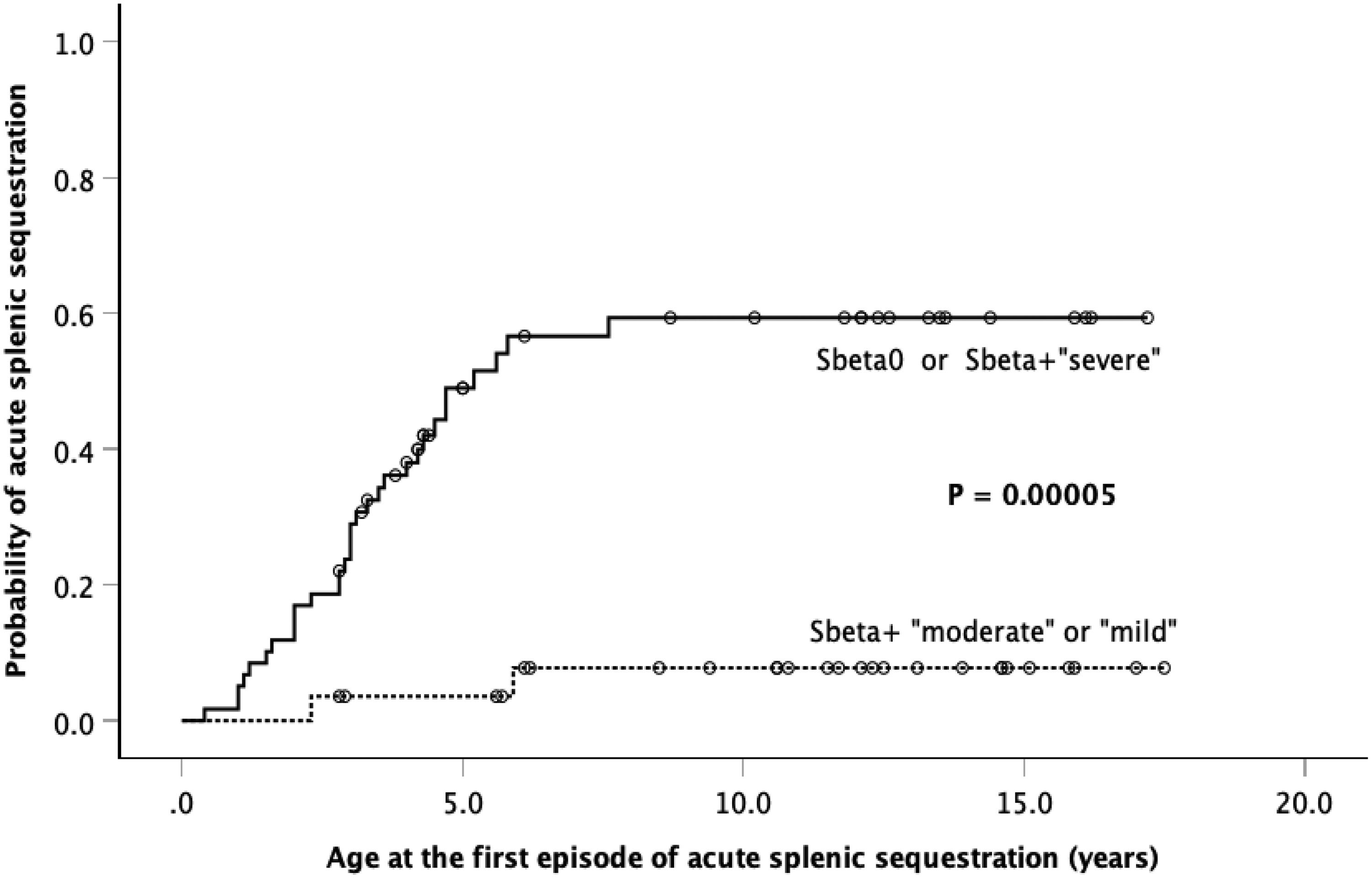
Including all children, 43 (48 %) had the spleen border at least 3 cm below the left costal margin. Out of these, 30 (69.8 %) had HbSβ0-thal and 13 (30.2 %), HbSβ+-thal (seven in eight children with IVS-I-110 G>A, four in ten with IVS-I-6 T>C, and two in six children with IVS-I-5 G>A mutations). Hypersplenism was observed in three children. Thirty-three children (37.1 %) had at least one episode of acute splenic sequestration (ASS). Age at first acute splenic sequestration ranged from 5 months to 7.6 years, and the cumulative probability was 41.2 % ± 5.7 %. Out of those 33 children, 25 (75.8 %) had HbSβ0-thal and eight (24.2 %), HbSβ+-thal (4/8 with IVS-I-110 G>A, 2/6 with IVS-I-5 G>A, and 2/10 with IVS-I-6 T>C, and mutations). Adding HbSβ0-thal to severe HbSβ+-thal and comparing with moderate and mild HbSβ+-thal, the cumulative probabilities of ASS were 59.3 %±7.2 % and 7.8 %±5.3 %, respectively (p = 0.00005; Figure 2).
![Kaplan-Meier probability curve [Plot (1-Survival)] for the occurrence of the first episode of acute splenic sequestration comparing children with “HbSβ0 or severe HbSβ+-thalassemia” and those with “moderate or mild HbSβ+-thalassemia”.](https://static.elsevier.es/multimedia/25311379/unassign/S2531137923025981/v1_202401050422/en/main.assets/gr2.jpeg?xkr=ue/ImdikoIMrsJoerZ+w92xcqUtYYCc6yiYRjTD2AR1aFJOKb7qLd+WBf18Ptk/O8dVG5SMqADZji5skBxdmL4+FHZiP/XNTrru3p+uK4F6cmFvHt71IJaDKLtMrd8fsjlo96iRli+WahT5e7q1/h6iYBSDMg1ZOk0hi1bn1nWOaWjodWBCp5A501zxQ4yG1EVyIn0XkTY3uCkGGS85Ta4d8Ghst08sXRBvp5/qqus2GLhMDRkJv2g8HytWiiIL7n12+awFoN7fKDKxvWq8dkyOPwHSHM2+KvHxVzm9HNKu99abGRXkQNQQXCV+H2j2l "Kaplan-Meier probability curve [Plot (1-Survival)] for the occurrence of the first episode of acute splenic sequestration comparing children with “HbSβ0 or severe HbSβ+-thalassemia” and those with “moderate or mild HbSβ+-thalassemia”.")
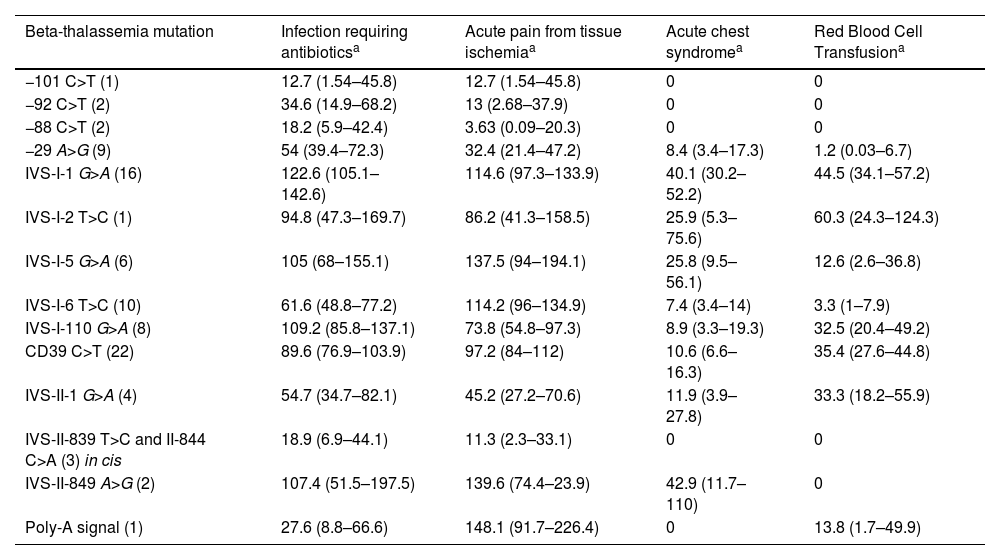
Seventy-seven (85 %) children had at least one acute vaso-occlusive crisis resulting in pain (VOCP), totaling 647 events during follow-up period. The incidence of VOCP was 83.6 events per 100 pt-yrs (95 % CI: 77.4 – 90.2). During the follow-up, 82 (94.3 %) children had at least one infection episode requiring antibiotic administration. The incidence of infection was 79 events per 100 pt-yrs (95 % CI: 72.9 – 85.4). Forty-eight (55.2 %) children had at least one episode of acute chest syndrome (ACS), as defined in [11]. The incidence of ACS was 14.5 events per 100 pt-yrs (95 % CI: 12.0 – 17.4). At least one red blood cell transfusion was necessary in 49 children (56.3 %). The incidence of transfusion was 14.5 events per 100 pt-yrs (95 % CI: 12.0 – 17.4). Incidence of clinical events of HbSβ-thal children according to β-thalassemia causative variants is depicted in Table 2.
Incidence of clinical events per 100 patient-years in children with HbSβ-thalassemia according to β-thalassemia mutation.
| Beta-thalassemia mutation | Infection requiring antibioticsa | Acute pain from tissue ischemiaa | Acute chest syndromea | Red Blood Cell Transfusiona |
|---|---|---|---|---|
| −101 C>T (1) | 12.7 (1.54–45.8) | 12.7 (1.54–45.8) | 0 | 0 |
| −92 C>T (2) | 34.6 (14.9–68.2) | 13 (2.68–37.9) | 0 | 0 |
| −88 C>T (2) | 18.2 (5.9–42.4) | 3.63 (0.09–20.3) | 0 | 0 |
| −29 A>G (9) | 54 (39.4–72.3) | 32.4 (21.4–47.2) | 8.4 (3.4–17.3) | 1.2 (0.03–6.7) |
| IVS-I-1 G>A (16) | 122.6 (105.1–142.6) | 114.6 (97.3–133.9) | 40.1 (30.2–52.2) | 44.5 (34.1–57.2) |
| IVS-I-2 T>C (1) | 94.8 (47.3–169.7) | 86.2 (41.3–158.5) | 25.9 (5.3–75.6) | 60.3 (24.3–124.3) |
| IVS-I-5 G>A (6) | 105 (68–155.1) | 137.5 (94–194.1) | 25.8 (9.5–56.1) | 12.6 (2.6–36.8) |
| IVS-I-6 T>C (10) | 61.6 (48.8–77.2) | 114.2 (96–134.9) | 7.4 (3.4–14) | 3.3 (1–7.9) |
| IVS-I-110 G>A (8) | 109.2 (85.8–137.1) | 73.8 (54.8–97.3) | 8.9 (3.3–19.3) | 32.5 (20.4–49.2) |
| CD39 C>T (22) | 89.6 (76.9–103.9) | 97.2 (84–112) | 10.6 (6.6–16.3) | 35.4 (27.6–44.8) |
| IVS-II-1 G>A (4) | 54.7 (34.7–82.1) | 45.2 (27.2–70.6) | 11.9 (3.9–27.8) | 33.3 (18.2–55.9) |
| IVS-II-839 T>C and II-844 C>A (3) in cis | 18.9 (6.9–44.1) | 11.3 (2.3–33.1) | 0 | 0 |
| IVS-II-849 A>G (2) | 107.4 (51.5–197.5) | 139.6 (74.4–23.9) | 42.9 (11.7–110) | 0 |
| Poly-A signal (1) | 27.6 (8.8–66.6) | 148.1 (91.7–226.4) | 0 | 13.8 (1.7–49.9) |
Incidence per 100 patient-years and 95 % confidence interval. Definition of clinical events according to.11
Data for groups with less than 5 children should be interpreted with caution.
Out of 89 HbSβ-thal children, one (1.1 %) had already had stroke (CD39 C>T) before starting the TCD screening in our center. Out of those 89, 68 (76.4 %) had at least one TCD test. Two children (IVS-I-5 G>A and IVS-I-110 G>A) had low conditional TCD and 66 (97.1 %) had low-risk TCD.
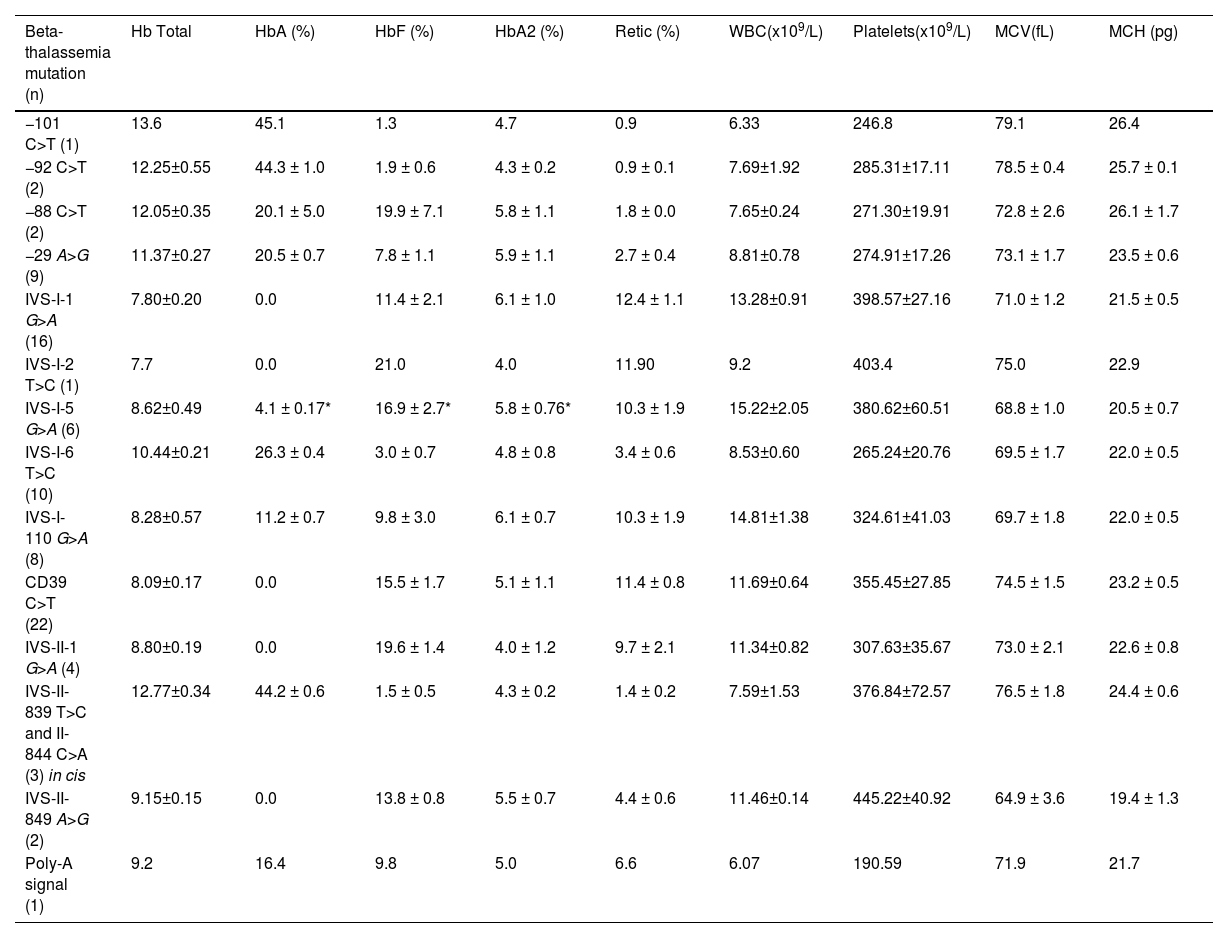
Laboratory profile of children with HbSβ-thalassemiaIn general, children with HbSβ-thal had low levels of MCH and MCV while HbA2 levels were high; there was no significant difference in levels of MCH, MCV, and HbA2 levels between individuals with HbSβ0-thal and HbSβ+-thal. Children with HbSβ0-thal had significantly lower total Hb levels and higher WBC, platelets, reticulocytes, and HbF when compared to those with HbSβ+-thal. Children with HbSβ+-thal presented a wide spectrum of HbA, ranging from 3.7 % (IVS-I-5 G>A) to 45.2 % (−92 C>T). Laboratory data of HbSβ-thal children according to β-thalassemia mutation are depicted in Table 3.
Baseline laboratory data of children with HbSβ-thalassemia according to β-thalassemia mutation.
| Beta-thalassemia mutation (n) | Hb Total | HbA (%) | HbF (%) | HbA2 (%) | Retic (%) | WBC(x109/L) | Platelets(x109/L) | MCV(fL) | MCH (pg) |
|---|---|---|---|---|---|---|---|---|---|
| −101 C>T (1) | 13.6 | 45.1 | 1.3 | 4.7 | 0.9 | 6.33 | 246.8 | 79.1 | 26.4 |
| −92 C>T (2) | 12.25±0.55 | 44.3 ± 1.0 | 1.9 ± 0.6 | 4.3 ± 0.2 | 0.9 ± 0.1 | 7.69±1.92 | 285.31±17.11 | 78.5 ± 0.4 | 25.7 ± 0.1 |
| −88 C>T (2) | 12.05±0.35 | 20.1 ± 5.0 | 19.9 ± 7.1 | 5.8 ± 1.1 | 1.8 ± 0.0 | 7.65±0.24 | 271.30±19.91 | 72.8 ± 2.6 | 26.1 ± 1.7 |
| −29 A>G (9) | 11.37±0.27 | 20.5 ± 0.7 | 7.8 ± 1.1 | 5.9 ± 1.1 | 2.7 ± 0.4 | 8.81±0.78 | 274.91±17.26 | 73.1 ± 1.7 | 23.5 ± 0.6 |
| IVS-I-1 G>A (16) | 7.80±0.20 | 0.0 | 11.4 ± 2.1 | 6.1 ± 1.0 | 12.4 ± 1.1 | 13.28±0.91 | 398.57±27.16 | 71.0 ± 1.2 | 21.5 ± 0.5 |
| IVS-I-2 T>C (1) | 7.7 | 0.0 | 21.0 | 4.0 | 11.90 | 9.2 | 403.4 | 75.0 | 22.9 |
| IVS-I-5 G>A (6) | 8.62±0.49 | 4.1 ± 0.17* | 16.9 ± 2.7* | 5.8 ± 0.76* | 10.3 ± 1.9 | 15.22±2.05 | 380.62±60.51 | 68.8 ± 1.0 | 20.5 ± 0.7 |
| IVS-I-6 T>C (10) | 10.44±0.21 | 26.3 ± 0.4 | 3.0 ± 0.7 | 4.8 ± 0.8 | 3.4 ± 0.6 | 8.53±0.60 | 265.24±20.76 | 69.5 ± 1.7 | 22.0 ± 0.5 |
| IVS-I-110 G>A (8) | 8.28±0.57 | 11.2 ± 0.7 | 9.8 ± 3.0 | 6.1 ± 0.7 | 10.3 ± 1.9 | 14.81±1.38 | 324.61±41.03 | 69.7 ± 1.8 | 22.0 ± 0.5 |
| CD39 C>T (22) | 8.09±0.17 | 0.0 | 15.5 ± 1.7 | 5.1 ± 1.1 | 11.4 ± 0.8 | 11.69±0.64 | 355.45±27.85 | 74.5 ± 1.5 | 23.2 ± 0.5 |
| IVS-II-1 G>A (4) | 8.80±0.19 | 0.0 | 19.6 ± 1.4 | 4.0 ± 1.2 | 9.7 ± 2.1 | 11.34±0.82 | 307.63±35.67 | 73.0 ± 2.1 | 22.6 ± 0.8 |
| IVS-II-839 T>C and II-844 C>A (3) in cis | 12.77±0.34 | 44.2 ± 0.6 | 1.5 ± 0.5 | 4.3 ± 0.2 | 1.4 ± 0.2 | 7.59±1.53 | 376.84±72.57 | 76.5 ± 1.8 | 24.4 ± 0.6 |
| IVS-II-849 A>G (2) | 9.15±0.15 | 0.0 | 13.8 ± 0.8 | 5.5 ± 0.7 | 4.4 ± 0.6 | 11.46±0.14 | 445.22±40.92 | 64.9 ± 3.6 | 19.4 ± 1.3 |
| Poly-A signal (1) | 9.2 | 16.4 | 9.8 | 5.0 | 6.6 | 6.07 | 190.59 | 71.9 | 21.7 |
Data shown as mean ± standard error of the mean (±SEM).
Data for groups with less than 5 children should be interpreted with caution.
Clinical and laboratory characteristics of patients according to beta thalassemia mutation are depicted in Tables 2 and 3, respectively. Laboratory profile was similar in children with CD39 C>T, IVS-I-1 G>A, IVS-I-5 G>A, or IVS-I-110 G>A causative variants, except for HbA2, and the degree of microcytosis and hypochromia (Supplementary Table 2). However, children with IVS-I-1 G>A mutation had significantly higher incidence of infection and ACS when compared to those with CD39 C>T (p<0.01), while children with HbSβ+-severe (IVS-I-110 G>A or IVS-I-5 G>A) had similar incidence of clinical outcomes when compared to those with CD39 C>T (p>0.05).
Children with CD39 C>T had a more severe laboratory profile when compared with those with −29 A>G or IVS-I-6 T>C, except for MCV and MCH values that were similar (Supplementary Table 3). Furthermore, children with −29 A>G or IVS I-6 T>C variants presented significantly lower incidence of infection and transfusion when compared to those with CD39 C>T (p<0.01), while children with −29 A>G also had significantly lower incidence of VOCP when compared to those with CD39 C>T (p<0.01).
Children with HbSβ+-intermediate had a more severe laboratory profile when compared with those with HbSβ+-mild (Supplementary Table 4). Furthermore, children with HbSβ+-intermediate had significantly higher incidence of infections (p = 0.001), VOCP (p<0.001), and ACS (p = 0.046) when compared to those with HbSβ+-mild. However, there was no significant difference in the incidence of transfusions between individuals with HbSβ+-intermediate and HbSβ+-mild (p = 0.39; Supplementary Table 4).
Effect of α-thalassemiaThe quantity or quality of genomic DNA was not sufficient for genotyping haplotypes and α-thal in one child (−29 A>G). About a fifth (n = 18/86; 20.9 %) of the children had coexistent a 3.7 kb HBA1/HBA2 deletion (16 αα/-α3.7, one -α3.7/-α3.7, and one αα/ααα−3.7). The analysis to assess the effect of α-thal on phenotype was restricted to children with the mutations CD39 C>T or IVS I-1 G>A because there was a higher number of individuals in these groups. There were no significant association between laboratory and clinical characteristics and coexistence of 3.7 kb HBA1/HBA2 deletion (data not shown).
Effect of beta-globin gene cluster haplotypesFrequency of βS- and βThal-haplotypes according to β-thalassemia mutation in 86 children with HbSβ-thalassemia is depicted in Table 4. The analysis to assess the effect of βS-haplotypes on phenotype was restricted to children with the mutations CD39 C>T. Children with the Benin haplotype had a higher incidence of infections and ACS than children with CAR (p = 0.04 and p = 0.03, respectively). There was no association between βS-haplotypes and transfusions or acute splenic sequestration in children with CD39 C>T.
Frequency of βS and βThal-haplotypes according to β-thalassemia mutation in 86 children with HbSβ-thalassemia.
| β-thalassemia mutation (n) | βS/βThal-haplotypes (n) |
|---|---|
| −101 C>T (1) | Ben/I (1) |
| −92 C>T (2) | CAR/V (2) |
| −88 C>T (2) | CAR/VII (1) and CAR/ATP (1) |
| −29 A>G TATA box (8) | Ben/IX (3), Sen/IX (2), Car/IX (1), CAR/I (1), Ben/ATP (1) |
| IVS-I-1 G>A (16) | CAR/V (8), Ben/V (6), CAR/I (2) |
| IVS-I-2 T>C (1) | CAR/ATP or Ben/ATP |
| IVS-I-5 G>A (6) | CAR/V (4, two siblings), Ben/V (1), ATP/V (1) |
| IVS-I-6 T>C (10) | CAR/VI (2), Ben/VII (2), CAR/IV (1), Ben/VI (1), CAR/?a (4) |
| IVS-I-110 G>A (8) | CAR/I (5), Ben/I (2), Ben/?a (1) |
| CD39 C>T (22) | CAR/II (11), Ben/II (2), CAR/?a (5), Ben/?a (4) |
| IVS-II-1 G>A (4) | CAR/V (1), Ben/V (1), CAR/ATP or Ben/APT (2 siblings) |
| IVS-II-839 T>C and IVS-II-844 C>A in cis (3) | CAR/ATP or Ben/ATP (3) |
| IVS-II-849 A>G (2) | CAR/ATP or Ben/ATP (2 siblings) |
| Poly-A signal (1) | CAR/I (1) |
The relative incidence of HbSβ-thal represents about 6 % of all cases with SCD in Minas Gerais. The incidence of HbSβ+-thal and that of HbSβ0-thal subtypes were almost the same.
To the best of our knowledge, the state of Minas Gerais is the most heterogeneous state in terms of β-Thal alleles in Brazil. The most prevalent allele found in the present study was CD39 C>T, corroborating previous studies carried out in the Southeastern region of Brazil.13,27-34 In the Mediterranean population, this variant accounts for the second most common one.4
In the present cohort, children with HbSβ+-thal presented a broad spectrum of clinical phenotypes, while children with HbSβ0-thal generally had a severe disease, as demonstrated by several reports.13
Overall, 14 different β-thalassemia mutations were identified in this study. While the type of β-thalassemia mutation clearly affected the clinical outcome of children with HbSβ-thal, co-inherited α-thal and βS-haplotypes had no clinical impact in the present study. The relative HbA concentration has a protective effect on the morbidity of HbSβ-thal, most probably due to decrease in polymerization, sickling of red blood cells and other subsequent pathophysiological effects.35
The mutations found in the present study were categorized into four distinct groups according to clinical severity. The first comprises all genotypes without HbA production (HbSβ0-thal). Individuals with the HbSβ+-thal genotype were categorized into severe, moderate, and mild forms. Individuals with the IVSI-110 G>A or IVS-I-5 G>A mutations, with a relative concentration of HbA lower than 10 %, were included in the group of severe HbSβ+ and had, particularly those with IVS-I-5 G>A,36 clinical features similar to those with HbSβ0-thal.13 Individuals with mutations - 29 A>G, −88 C>T, IVS-I-6 T>C and in the polyadenylation region, with HbA concentrations between 16 % and 26 %, were included in the moderate HbSβ+ group. Children with −101 C>T, −92 C>T, and IVS-II double mutation in cis had HbA above 40 % and a very mild disease.
In this study, the prevalence of splenomegaly was 31 %, similar to other previous studies.5,37 Prevalence of splenomegaly was higher in children with the HbSβ0-thal and severe HbSβ+-thal (group 1) when compared to those with moderate or mild HbSβ+-thal (group 2). Correspondingly, group 1 had more acute splenic sequestration than group 2. Therefore, low HbA levels in the severe forms of HbSβ+-thal do not exempt these patients from having splenic events similar to those with HbSβ0-thal. This finding may be used to target those individuals at highest risk for the occurrence of splenic crises in educational and prevention policies for SCD.
Evaluation of clinical manifestations in children with different β-Thal mutations generally revealed that the incidence of clinical events per 100 patient-years was similar in those with HbSβ0-Thal and with IVS-I-5 G>A or IVS-I-110 G>A genotypes. These data can be explained by the low concentration of HbA observed in both β+-Thal mutations, as observed by others38,39 and by our group.36
In general, the clinical picture of HbSβ0-thal children with different mutations was similar, as expected from the absence of Hb A production. When they are compared to homozygous Hb SS patients, similarities are prominent40 and traditionally Hb SS and HbSβ0-thal are merged into a clinical entity named sickle cell anemia. However, differences do exist. For instance, cerebrovascular events are much less common in children with HbSβ0-thal when compared to those with Hb SS.41 Only a single case of overt stroke in a child with CD39 C>T was observed in the present cohort. As stated in Results, mild abnormalities in TCD were present in only two children with severe HbSβ+-thal.
Although the −29 TATA box mutation is often related to general mild clinical presentation, it has been associated in other studies with more severe clinical outcomes related to blood viscosity.3,4 Although some important differences in the studies hinder direct comparisons, our data corroborate these previous studies since VOCP and ACS were common clinical manifestations observed in children with −29 TATA box. Out of nine children with this mutation, five (55.6 %) had at least one ACS and an average of 2 ± 3.4 episodes of ACS was recorded in the follow-up period. These findings are important to guide clinical management in patients with this allele.
The effects of co-inheritance of α-thal in individuals with HbSβ-thal are still not well understood and there are controversies in the literature. Some studies showed a strong modulation of α-thal in HbSβ-thal phenotype,42,43 while other studies did not find significant influence.10,39,44 We have previously observed influence of α-thal in clinical and laboratory characteristics of children with HbSS and HbSC,8,9 suggesting an impact of α-thal in morbidity of SCD in our population. However, co-inheritance of α-thal had no significant influence on the laboratory and clinical parameters of children with the most frequent mutations in this study (IVS I-1 G>A and CD 39 C>T). Further studies are thus needed to understand the influence of co-inheritance of α-thal on the phenotypes of individuals with HbSβ-thal.
During Brazil colonization, the main people brought by slave ships came from the regions of Bantu (CAR) and Benin.37 This is corroborated with higher frequency of the CAR (57.5 %) and Benin (28.7 %) βS-haplotypes observed in this study. In addition, other studies carried out in the state of Minas Gerais also detected a high frequency of the CAR and Benin haplotypes in individuals with HbSS and HbSC.8,41 βS-haplotypes were postulated to influence the severity of SCD by influencing the concentration of HbF. However, there is no evidence to suggest that βS-haplotypes give any more information than measuring the HbF level.7 Although we observed a higher incidence of infections and ACS in children with Benin βS-haplotype when compared to those with CAR, the clinical impact is not relevant, making it unfeasible to use the βS-haplotypes as a marker for HbSβ-thal severity. Our findings do not support the hypothesis that the CAR βS-haplotype leads to a more severe phenotype. We previously observed no difference in the phenotypic characteristics of individuals with the CAR and Benin haplotypes in our cohorts of HbSS and HbSC children.8,41
The most frequent βthal-haplotypes in the present study were V (27.6 %), I (14.9 %) and II (14.9 %), and were identified mainly with β-thal alelles of European origin, such as IVS-I-110 G >A and CD39 C>T. However, some βthal-haplotypes found in this study are from African origin, such as those identified in the −29 C>T and in −88 C>T mutations. βthal-haplotypes are not restricted to a single β-thal mutation and have a heterogeneous distribution.45.
There are some limitations of this study. First, it has a retrospective design and missing data in medical records are possible. Second, given that the participants are children, long-term complications were not evaluated. Finally, the low frequency of some β-thal mutations as well as the low frequency of βS-haplotypes and α-thalassemia within each β-thal mutation are limiting factors for a more accurate evaluation of genotype-phenotype correlation.
ConclusionsThe incidence of HbSβ-thal in the Brazilian state of Minas Gerais is 1 per 22,250 newborns. Laboratory parameters and incidence of clinical manifestations are quite similar in children with HbSβ0-thal genotypes and severe HbSβ+-thal, while infants with HbSβ+-thal intermediate or mild had a varying clinical presentation. We have shown that the underlying β-thalassemia mutation was associated with clinical and laboratory features in children with HbSβ-thal, while βS-haplotypes and α-thalassemia did not meaningfully influence the phenotype. The early identification of β-thalassemia mutation may help the clinical management of children and lead to a better prognosis and survival of children with HbSβ-thal.


