

Sickle cell disease (SCD) results from a single amino acid substitution in the gene encoding the β-globin subunit (β6Glu>Val) that produces the abnormal hemoglobin (Hb) named Hb S. SCD has different genotypes with substantial variations in presentation and clinical course (Table 1).1,2 The combination of the sickle cell mutation and beta-thalassemia (β-Thal) mutation gives rise to a compound heterozygous condition known as Hb S/β thalassemia (Hb S/β-Thal), which was first described in 1944 by Silvestroni and Bianco.3
Sickle cell disease genotypes.
| Severe sickle cell disease | ||
| Sickle cell anemia (Hb S/S) | β6Glu>Val/β6Glu>Val | The most common form |
| Hb S/β0 thalassemia | Multiple mutations | Most prevalent in the eastern Mediterranean region and India |
| Severe Hb S/β+ thalassemia | Multiple mutations | Most prevalent in the eastern Mediterranean region and India |
| Hb S/O Arab | β6Glu>Val/β121Glu>Lys | North Africa, the Middle East, and the Balkans |
| Hb S/D Punjab | β6Glu>Val/β121Glu>Gln | Predominant in northern India |
| Hb S/C Harlem | β6Glu>Val/β6Glu>Val/β, β73Asp>Asn | Very rare |
| Moderate sickle cell disease | ||
| Hb S/C | β6Glu>Val/β6Glu>Lys | 25–30% cases of sickle cell disease of African origin |
| Moderate Hb S/β+ thalassemia | Multiple mutations | Most in the eastern Mediterranean region |
| Mild sickle cell disease | ||
| Mild Hb S/β++ thalassemia | Multiple mutations | Mostly in populations of African origin |
| Hb S/E | β6Glu>Val/β26Glu>Lys | Hb E predominates in southeast Asia |
| Very mild sickle cell disease | ||
| Hb S/Hereditary Persistence of Fetal Hemoglobin | Large deletions of the β-globin gene complex | |
The polymerization of deoxygenated Hb S (sickling) is the primary event in the molecular pathogenesis of SCD. However, this event is highly dependent on the intracellular Hb composition; in other words, it is dependent on the concentration of Hb S, and type and concentration of the other types of Hb. Therefore, the major primary genetic determinant of the severity of SCD is the genotype.2,4–6
Many different β-Thal mutations have been associated with Hb S, and the molecular basis of the thalassemia in Hb S/β-Thal individuals reflects the spectrum of β-Thal mutations observed in a particular population.5,7–12 The heterogeneity of the β-Thal mutations leads to quantitatively different β-globin synthesis and consequently to different amounts of Hb A. This fact results in variable clinical manifestations, ranging from nearly asymptomatic to a severe condition similar to sickle cell anemia (homozygous Hb S).3,13 There is no consensus about the classification of Hb S/β-Thal, but it is usually classified in two types: Hb S/β0-Thal and Hb S/β+-Thal.2,4
Hb S/β0-Thal, in which the production of Hb A is abolished, is often clinically indistinguishable from sickle cell anemia. The thalassemia acts on sickled red blood cells, inducing microcytosis, hypochromia, and sometimes Hb F is elevated. This result in an improvement of the circulatory competence of these cells, a reduction of hemolysis, and a small increase in Hb concentration and in packed cell volume. However, these effects are not accompanied by any reduction in vaso-occlusive events, probably due to the great number of Hb S-containing red blood cells resulting in increased blood viscosity.4,5
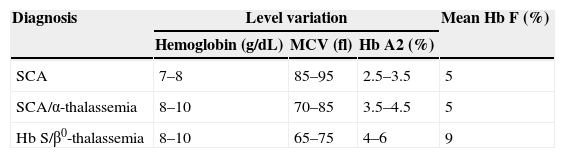
A confusing diagnostic problem is the differentiation of Hb S/β0-Thal from sickle cell anemia associated with α-thalassemia. Table 2 shows that hematologic and electrophoretic studies are unable to distinguish between the two conditions and so family studies and DNA analysis are needed to confirm the diagnosis.5
Laboratory differentiation of sickle cell anemia, sickle cell anemia/α-thalassemia, and Hb S/β0-thalassemia.
| Diagnosis | Level variation | Mean Hb F (%) | ||
|---|---|---|---|---|
| Hemoglobin (g/dL) | MCV (fl) | Hb A2 (%) | ||
| SCA | 7–8 | 85–95 | 2.5–3.5 | 5 |
| SCA/α-thalassemia | 8–10 | 70–85 | 3.5–4.5 | 5 |
| Hb S/β0-thalassemia | 8–10 | 65–75 | 4–6 | 9 |
SCA: sickle cell anemia; MCV: mean corpuscular volume; Hb A2: hemoglobin A2; Hb F: hemoglobin fetal.
In Hb S/β+-Thal, variable amounts of Hb A dilute Hb S and consequently inhibit polymerization-induced cellular damage. The Hb A levels vary from <5% to 45% of the hemolysate and higher levels of Hb A are usually associated with a milder phenotype.5,8,12–15 However, because of the confounding influences of other genetic modifiers, such as γ-globin gene expression and α-thalassemia, a rigid genotype–phenotype correlation is difficult to establish.5
A classification of Hb S/β+-Thal into Types I (Hb A: 1–7%), II (Hb A: 7–14%), or III (Hb A: 14–25%) according to the level of Hb A has been proposed.3,14 However, this classification is not widely accepted and could be considered of little utility. Nowadays it is possible to define the β-Thal mutation exactly by molecular biology techniques, and determine the relationship between mutation and clinical manifestations.5
The determination of the β-Thal mutation in Hb S/β+-Thal individuals was performed by Besilario et al. These authors studied four patients with an Hb A concentration above 38% and a very mild form of the disease, and identified two mutations for the first time in the Brazilian population.16 It is important to point out that these cases could be wrongly interpreted as sickle cell trait, since they had no anemia, despite a little microcytosis, and that this incorrect diagnosis would reflect in the genetic counseling provided. In summary, this study reinforces the importance of molecular studies in our population, in order to enhance our knowledge about the disease in Brazil and consequently improve genetic counseling, follow up, and treatment.
Conflicts of interestThe author declares no conflicts of interest
The author wishes to thank Coordenação de Aperfeiçoamento de Pessoal de Nivel Superior (CAPES) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) for financial support.
See paper by Belisário et al. on pages 198-201.


