
Some autoimmune and chronic inflammatory disorders are associated with an increased risk of Non-Hodgkin's Lymphoma (NHL). Despite recent extensive research, the causes of NHL remain poorly understood. The most common pathologies related to NHL are rheumatoid arthritis, Sjögren's syndrome, systemic lupus erythematosus, celiac disease, dermatitis herpetiformis and chronic thyroiditis.1 Still's disease (SD) is a rare inflammatory disorder which mostly affects young people. The association between SD and lymphoma is rare in the literature because their distinction is still difficult.2 We report here the case of a woman who was diagnosed with SD and developed NHL 12 months after the onset of the symptoms. To our knowledge, this is the sixteenth reported case of such an occurrence.
Case reportA 48-year-old female, previously healthy, presented in September 2019 with fever, weight loss, adenomegaly and splenomegaly, accompanied by polyarthritis of the large joints. The computed tomography (CT) revealed axillary adenomegaly, associated with significant splenomegaly. A lymph node biopsy was performed more than once and the immunohistochemistry panel (IHC) was consistent with a reactive lymphadenopathy. Excluding malignancy, an evaluation by a rheumatologist confirmed the SD diagnosis, so treatment with methotrexate and tocilizumab was instituted.
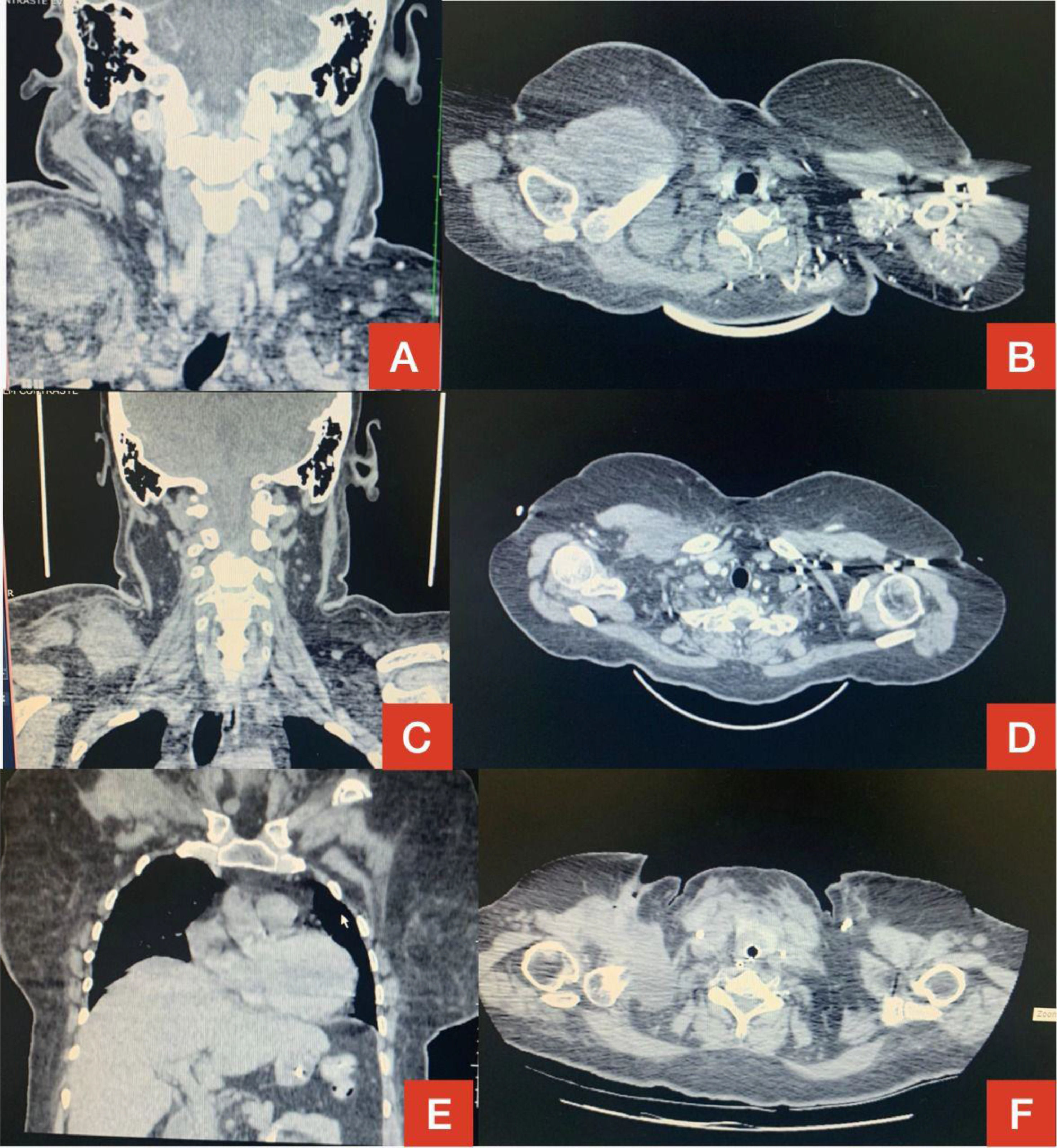
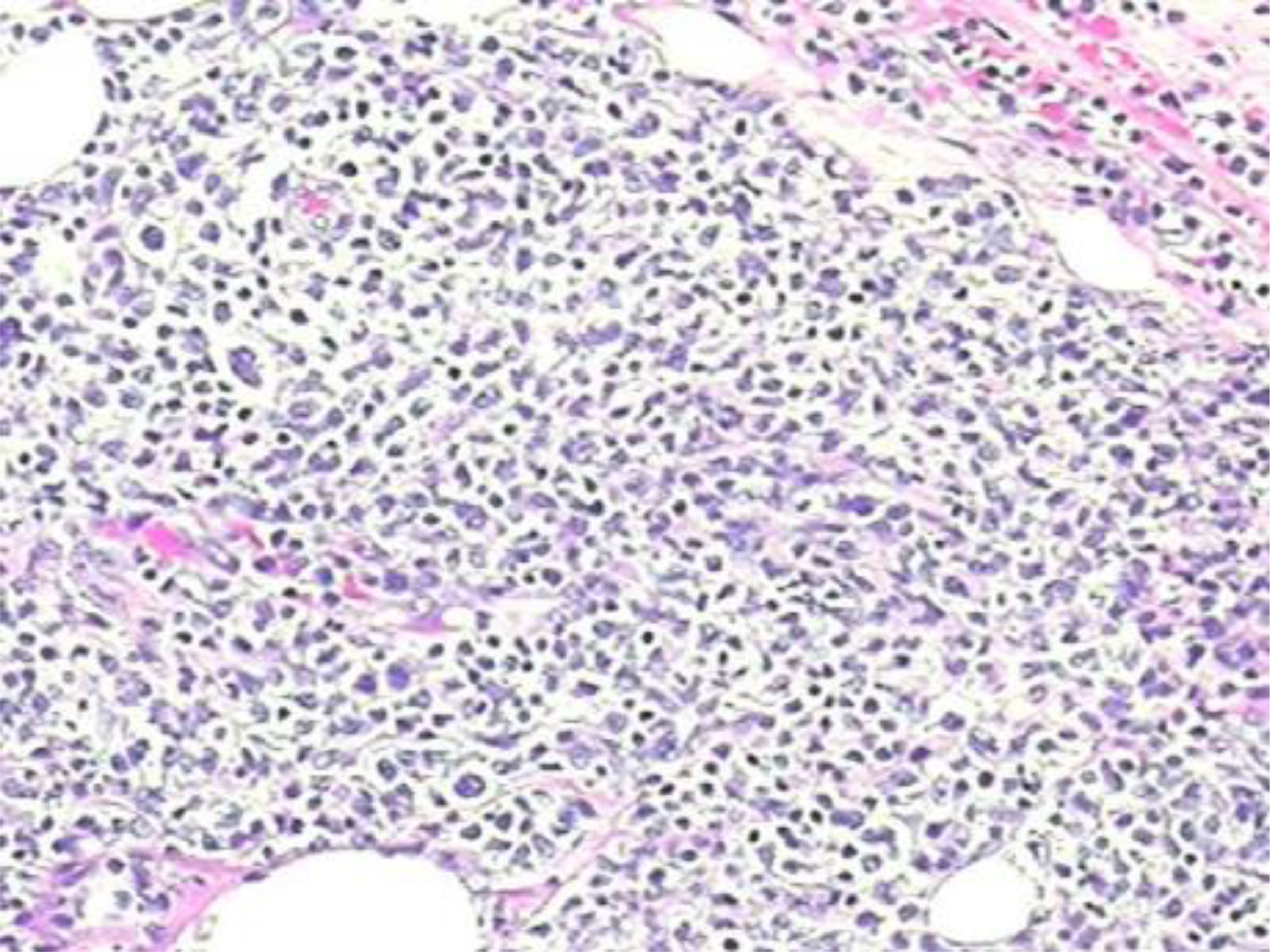
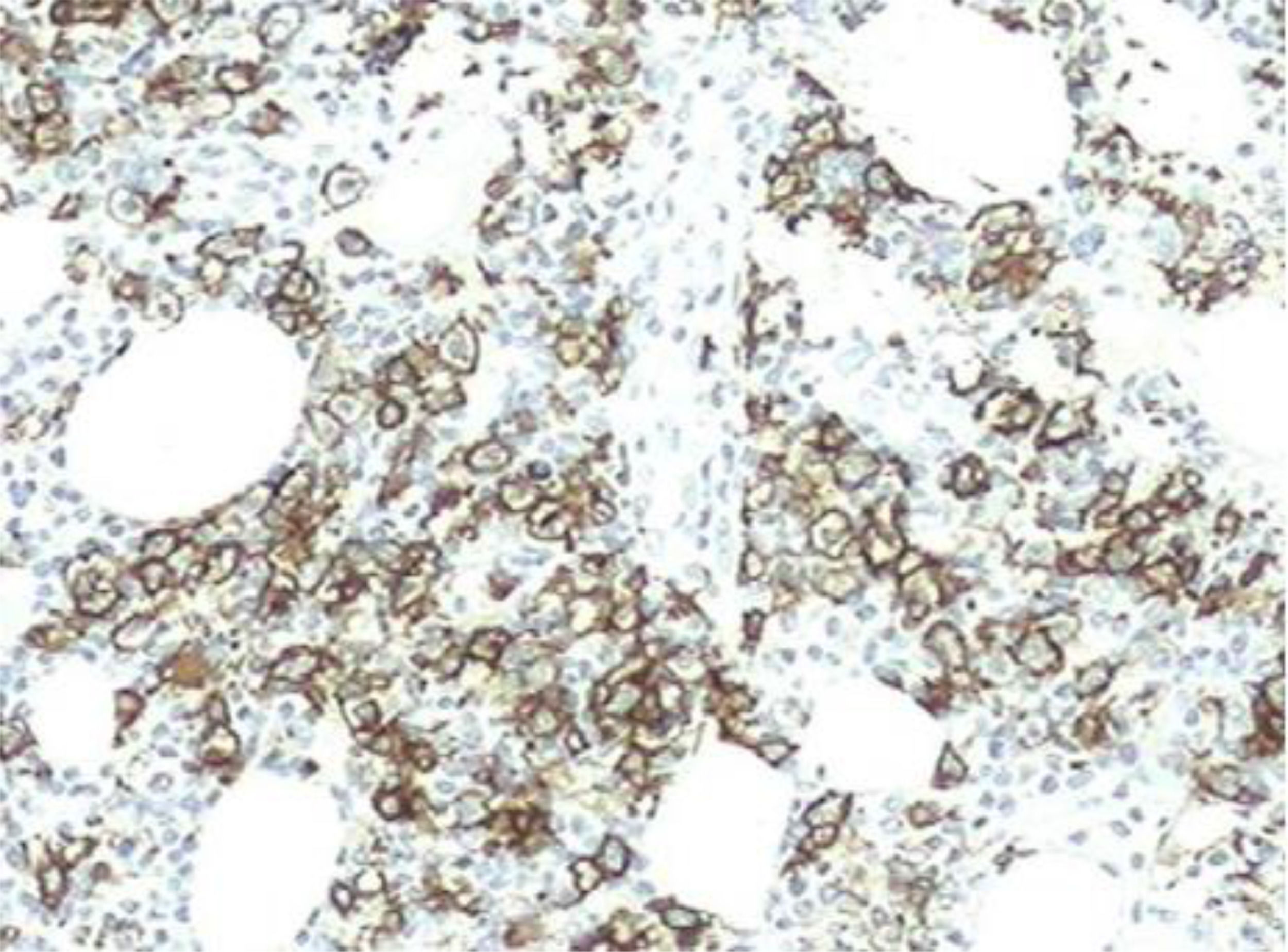
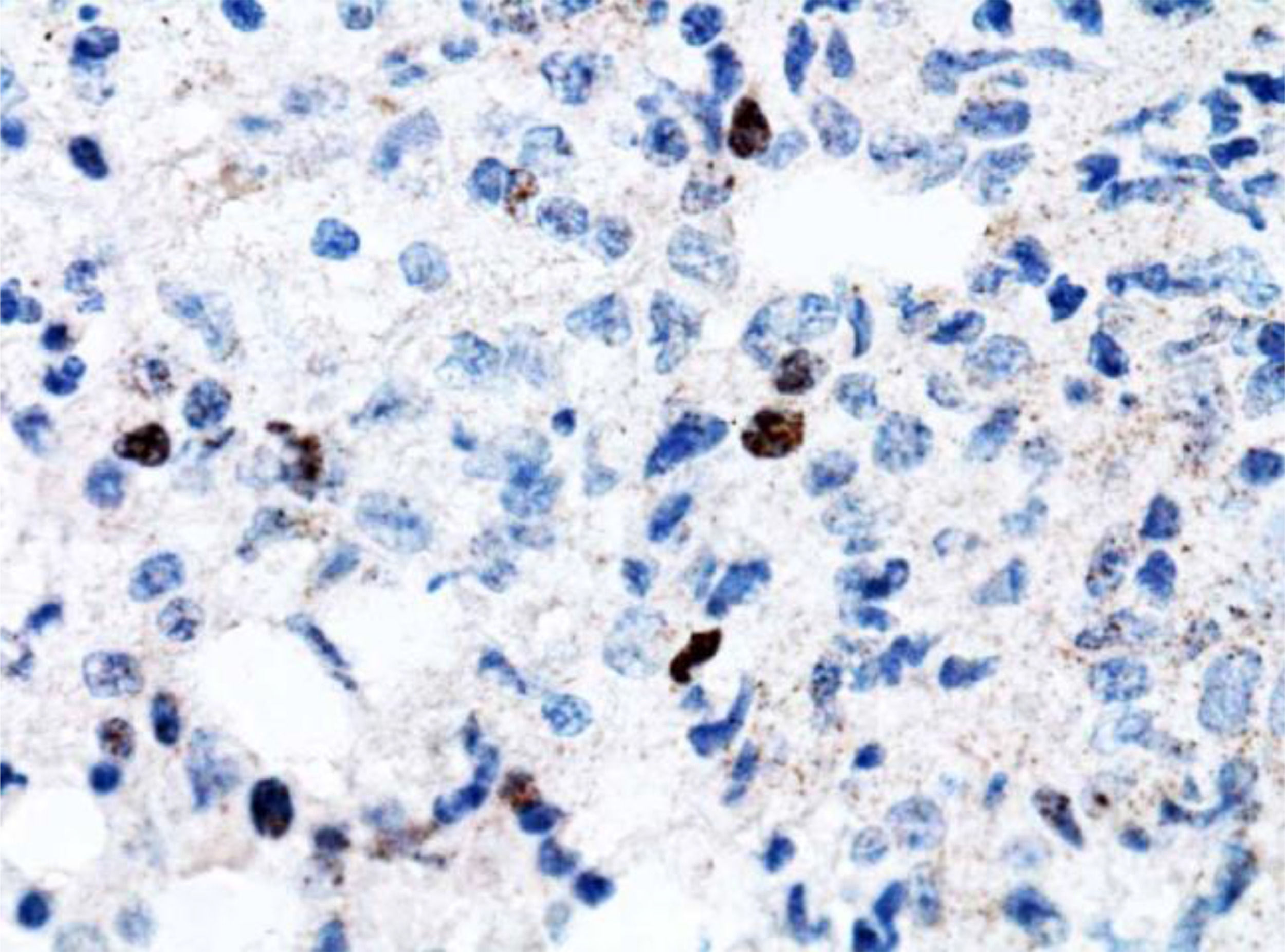
However, 12 months later, a right anterior chest wall lesion with local skin infiltration and inflammatory signs (redness, heat and pain) appeared, associated with fever, weight loss, and night sweats. The initial suspicion was an abscess, so a biopsy and a thoracic drainage were performed. The rheumatologic treatment was discontinued and, within a few weeks, there was a huge enlargement of the lesion. The CT demonstrated multiple cervical, axillary, retropectoral and mediastinal lymph nodes and the presence of a large solid lesion in the right pectoral and supra scapular region, embracing the clavicle and extending to the axillary region, measuring 10.4 × 7.6 cm (Figure 1). The immunohistochemistry diagnosis was difficult, but, after evaluation by the FISH (fluorescence in situ hybridization) test, a diagnosis of diffuse large B-cell lymphoma was confirmed (CD20 +, association with EBV +, CD3 - and CD79a +) (Figures 1,2 and 4). The bone marrow biopsy and CSF immunophenotyping showed no infiltration by lymphoma. At diagnosis, ECOG 1, IPI 2 and CNS-IPI 1 were observed.
 of the neck in coronal section (A) and CT of the chest in sagittal section (B) showing extensive lesion in the right side in December 2020. Reduction of tumor mass in January 2021 demonstrated on the same CT scans in C and D. Disease progression in March 2021 on chest CT in coronal (E) and sagittal (F) sections.")
Computed tomography (CT) of the neck in coronal section (A) and CT of the chest in sagittal section (B) showing extensive lesion in the right side in December 2020.
Reduction of tumor mass in January 2021 demonstrated on the same CT scans in C and D. Disease progression in March 2021 on chest CT in coronal (E) and sagittal (F) sections.

The R-CHOP chemotherapy was initiated and, after the first cycle, the patient had clinical worsening, fever, drowsiness, hypohemoglobinemia (5,8 g/dL), thrombocytopenia (18.000/mm3), leucopenia 2.260/mm3, hyperferritinemia (980 ug/l), elevation of transaminases (AST 247,4 U/l; ALT 405 U/l) and hypertriglyceridemia (402 mg/dL), with no evidence of an active infectious process. The patient developed pancytopenia on the second day after chemotherapy. Thus, a high probability of SD reactivation and secondary macrophage activation syndrome (MAS) was raised. Also, there was an increased hemophagocytic activity in the bone marrow. Pulse therapy (methylprednisolone) was initiated according to the rheumatological evaluation, with clinical improvement and resolution of the symptoms. There was a significant reduction in the chest wall lesion after the second chemotherapy cycle; however, unfortunately, before the third chemotherapy cycle, the disease progressed, with a large increase in the lesion, associated with local compressive symptoms (Figure 1). The rescue treatment was initiated with the R-ICE protocol, in association with the autologous bone marrow transplantation plan. After chemotherapy, the patient progressed with febrile neutropenia, a new SD reactivation, pulmonary septic shock due to multiresistant bacteria and systemic candidemia. She was transferred to the intensive care unit (ICU), with orotracheal intubation and intensive measures, but without clinical response, even with optimized treatment, and the patient died a few weeks later.
DiscussionWe describe the case of a woman with SD who was diagnosed with lymphoma 12 months after the onset of the symptoms. The patient fulfilled the Yamaguchi et al. criteria for SD.3 In this case, there is a possibility of a latent lymphoma, as the onset of the clinical course and lymph node biopsy was negative for lymphoma on more than one occasion. According to a database review in MEDLINE, 15 proven cases of SD associated with non-Hodgkin's lymphoma were identified, such as the case of our patient.
The SD in an adult, described by Eric Bywaters in the 70′s,4 is a rare multigenic autoinflammatory disease with a generally good prognosis,2 whose etiology remains uncertain.4 Its diagnosis is based on the combination of clinical (night fever, evanescent salmon maculopapular rash and arthralgias or arthritis, polyadenopathy and pharyngeal pain) and biological signs (hyperleukocytosis, hyperferritinemia and collapsed glycosylated ferritin) and the exclusion of autoimmune ferritin, infectious or neoplastic pathology.2,4
The SD associated with neoplasms usually satisfies Yamagushi's diagnostic criteria. However, some atypia should be alarming about the possibility of an underlying neoplasm, such as: unusual age, sudden symptoms onset, poor response to corticosteroid or immunosuppressive therapy, or laboratory abnormalities (cytopenia and significant hypergammaglobulinemia).2,5
The macrophage activation syndrome (MAS), on the other hand, is a feared complication of rheumatologic diseases; it is most often seen in the young population, in systemic juvenile idiopathic arthritis (JIA), and can also be seen in adult SD.6 The MAS is classified as primary or secondary. Primary MAS occurs in the context of genetic diseases; secondary MAS is associated with infections, malignant diseases (especially lymphomas), rheumatologic conditions and immunodeficiency.7 This syndrome is characterized by a dysfunctional immune response, which leads to an exacerbated expansion and activation of cytotoxic cells with hypersecretion of pro-inflammatory cytokines, resulting in hematological changes and organ damage.6 The mortality rate is between 10 and 22%.7 The main presentation is fever and other manifestations may include hepatosplenomegaly, lymphadenopathy, central nervous system (CNS) dysfunction and hemorrhages. Typical laboratory features are pancytopenia, hypofibrinogenemia, elevated liver enzymes, hypertriglyceridemia and elevated ferritin levels. Hemophagocytosis in the bone marrow or other reticuloendothelial tissues can also be identified.6
Lymphadenopathy commonly occurs in SD. Although most histopathological studies have shown non-diagnostic reactive hyperplasia in SD, histological patterns simulating malignant lymphoma have been reported. In addition to necrotizing lymphadenitis (Kikuchi disease8), some authors have described a distinct, intense paracortical hyperplasia characterized by immunoblastic cells and prominent arborizing vessels expansion. The intensity of this process, the erasure of apparent nodal architecture and atypical proliferative cells,4 combined with paracortical cell proliferation, apparent nodal involvement and atypical immunoblasts8 can suggest a misdiagnosis of malignancy. It may sometimes be difficult to draw a distinction from malignant hematologic disorders, not only because of the clinical features, but also because the histopathology of the lymph node biopsy may mimic lymphoma.4
In relation to the treatment, there is still no preconized protocol. The R-CHOP scheme stands out as the first line in NHL, as it requires combined chemotherapy. In case of refractory treatment, the protocol standard should be carefully reevaluated. Radiotherapy is appropriate for patients with local recurrence. There is no established consensus on bone marrow transplantation.9 (Figure 3)

 infected B lymphocytes.")
The consent for experimentation was obtained from the patient, observing privacy rights.


