
Hemophagocytic lymphohistiocytosis comprises a systemic hyperactivation of macrophages that requires prompt recognition of symptoms and early treatment.
Objective and MethodIn this context, we described clinical and laboratory characteristics, therapeutic modality and outcome of 21 patients with HLH treated at a pediatric oncology hospital between January 2000 and February 2019.
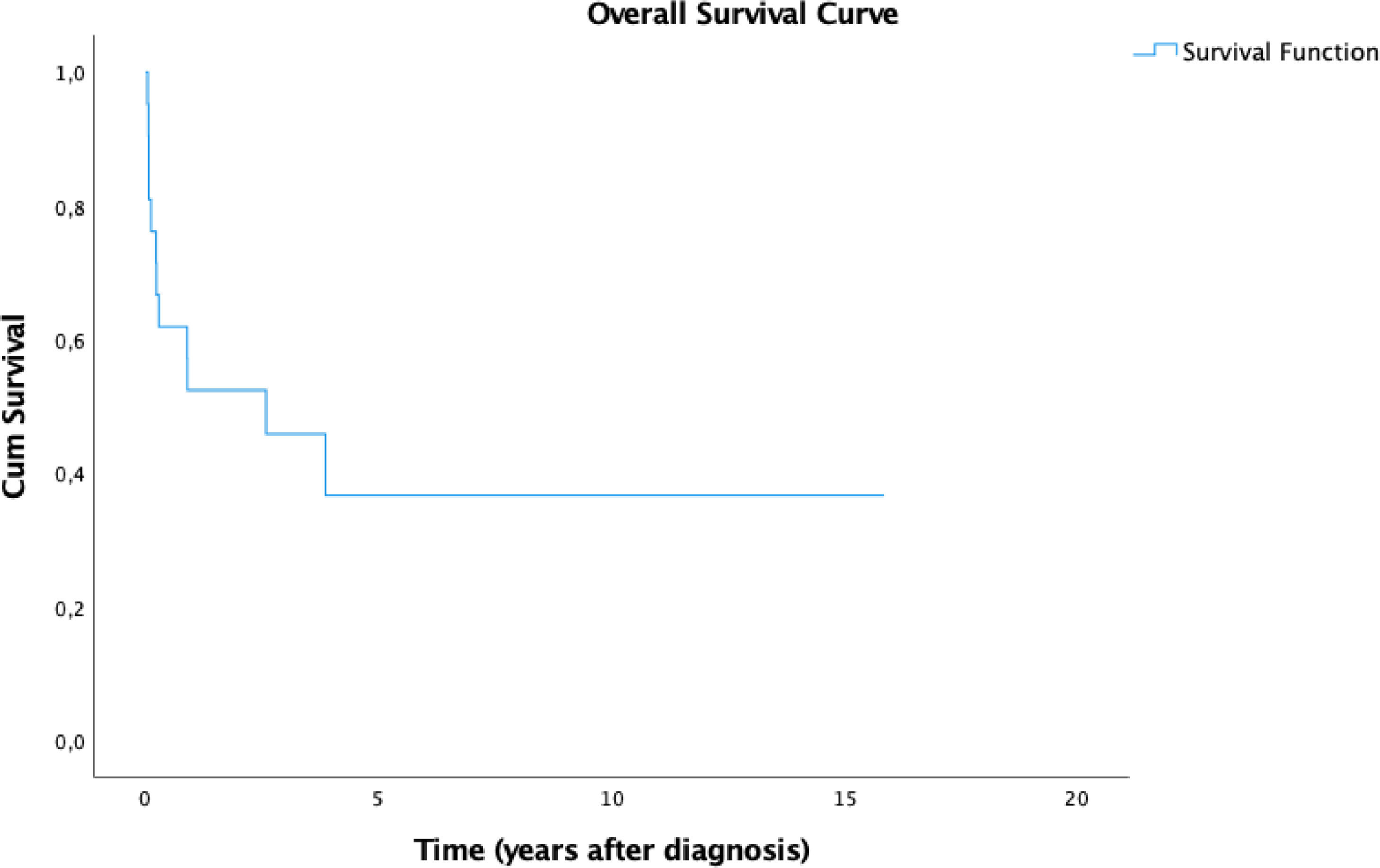
ResultsHLH mainly affected females, fever was the most frequent clinical sign and hyperferritinemia was the most prevalent laboratory abnormality. All patients were admitted to the intensive care unit (ICU) at some point. Fifteen (71.4%) patients presented resolution criteria and eight (53.3%) of them presented reactivation. The mortality rate was 57.1% and the mean time between diagnosis and death was 9.98 months. The 5-year overall survival (OS) was 36.7%. We observed a significant difference in prognosis associated with reactivation of HLH. These patients demonstrated an estimated 5-year OS of 25%, while all patients that did not reactivate were alive until the end of the follow-up.
ConclusionIn conclusion, HLH is a rare disease with a high mortality rate, especially in patients with disease reactivation and those with familial- or immunodeficiency-associated forms, which makes early recognition and genetic testing crucial for appropriate management and prompt SCT indication.
Hemophagocytic lymphohistiocytosis (HLH), also known as hemophagocytic syndrome, is a condition in which there is an inappropriate immune system activation. HLH comprises two different groups based on various inherited or acquired immune deficiencies.1
The familial form is rare and it underlies eight autosomal recessive gene defects that encode proteins related to the exocytosis of cytotoxic granules during apoptosis in natural killer (NK) cells.1,2 The acquired form is more frequent than the familial forms and it is a result of disrupted function of the immune system, as a consequence of severe infection, immunodeficiency or malignancy.3,4. The main histopathological characteristic of HLH is the infiltration of lymphocytes and histiocytes with hemophagocytic activity observed in the reticuloendothelial system, bone marrow and central nervous system.5 This systemic infiltration can explain the vast array of clinical manifestations.
Albeit rare, HLH is a potentially fatal condition. The full complex of HLH symptoms reflects an inability to control inflammatory responses. The cardinal manifestations of HLH are prolonged fever, pancytopenia and hepatosplenomegaly. 3,6 According to The Histiocyte Society (2004), the diagnosis is defined upon the presence of at least five of the following eight criteria: fever, splenomegaly, cytopenias affecting at least two of three lineages in the peripheral blood, hypertriglyceridemia and/or hypofibrinogenemia, hemophagocytosis in the bone marrow, spleen, or lymph nodes, low or absent NK‐cell activity, hyperferritinemia and high levels of sIL‐2r. Hemophagocytosis is characterized by the presence of erythrocytes, platelets or white blood cells at the macrophage cytoplasm.6
The treatment of HLH varies between the different subtypes of this disease and the outcome often depends on how quickly the diagnosis is recognized. In the case of familial HLH, the treatment usually involves immunochemotherapy followed by stem cell transplantation (SCT). Meanwhile, for secondary HLH, as used in the HLH-94 and HLH-2004 regimen, the treatment contains a combination of cyclosporin A, etoposide and corticosteroids.6,7
In this retrospective study, we describe the clinical features, laboratory findings and outcome of children with HLH at a single institution over a period of 19 years.
MethodsWe conducted a descriptive and analytical retrospective study of all the children with HLH admitted to the GRAACC/IOP/UNIFESP from January 2000 to February 2019. These patients were diagnosed using the current Histiocyte Society (2004) diagnostic criteria for HLH. For the descriptive analysis of categorical variables, frequency and percentage were calculated, and for continuous variables, the average, standard deviation, median, minimum and maximum. Survival curves were adjusted by the Kaplan-Meier. To compare the survival curves for cytopenia, hypertriglyceridemia, hypofibrinogenemia, central nervous system (CNS) involvement, SCT, hemoglobin, neutrophils, platelets and age, the logrank or Breslow tests were used. The Hazard Ratio was obtained from the Cox Simple Regression model. A significance level of 5% (p-value < 0.05) was used.
ResultsA total of 21 HLH cases were diagnosed from January 2000 to February 2019. Regarding epidemiologic data (Table 1), twelve patients were female and nine were male, with a sex ratio of 1:1.33. The age at diagnosis ranged from one month to 15 years, with a mean of 5.89 years (standard deviation 5.24).
Patient's general and epidemiological characteristics.
Seven of 21 patients had primary HLH: two associated with immunodeficiencies (one with ataxia telangiectasia and the other with the Chediak-Higashi syndrome), three who had the familial form (one with type 2 (mutation of the perforin gene - PFR1), one with type 3 (mutation of the UNC13D gene) and the other with type 5 HLH (mutation of the STXBP2 gene)). One patient was considered primary HLH because of consanguineous parents and another due to having two deceased brothers with HLH. Eight patients had an acquired form of HLH: one of them secondary to visceral leishmaniosis and whose clinical picture reversed with the treatment of this infection. The other cases of secondary HLH were associated with chemotherapy for choroid plexus carcinoma; after liver transplantation due to alpha-1-antitrypsin deficiency; after fungal peritonitis in a patient with chronic non-progressive encephalopathy; one patient had chronic renal disease with an EBV infection; one patient after scarlet fever; two patients did not present a clear cause for HLH and tested negative for familial mutations. The remaining six cases were not investigated for genetic mutation and, although some patients had a well-documented infection, this may have worked as a triggering factor in a child with a possible underlying genetic alteration.
All patients presented fever at diagnosis. Splenomegaly was found in nineteen patients, corresponding to 92.3%, and five patients presented hepatic involvement (characterized by coagulogram changes, cholestasis and elevated transaminases). Seven patients had involvement of the central nervous system (53.8%); such involvement was characterized by seizures, headache and a decreased level of consciousness. Acute lung edema and renal failure were seen in two other patients and mucosal hemorrhage was also observed in one. The most common laboratory finding was hypertriglyceridemia. Table 2 summarizes clinical and laboratory characteristics.
Clinical and laboratory data and outcomes of twenty-one patients with HLH.
P: patient; 1: primary; 2: secondary; 3: not tested; F: fever; S: splenomegaly HB: hemoglobin; LEU: leukocytes; NEU: neutrophils; PLA: platelets; TGL: hypertriglyceridemia; FIB: hypofibrinogenemia; HMF: hemophagocytosis; FER: ferritin; CNS: central nervous system.
Treatment and outcome.
The minimal time between the diagnosis and treatment was one day, while the maximum time was 10 days. The mean time between first symptoms and diagnosis was 25 days, varying from 4 to 124 days. The protocols used were HLH-94 in four patients (19%) and HLH-04 in 16 patients (76.1%). Only the patient with hemophagocytic syndrome (HPS) secondary to visceral leishmaniosis did not follow a protocol. Four patients (19%) underwent hematopoietic SCT. Twenty patients were admitted to the intensive care unit (ICU) at some point. Fifteen patients presented resolution criteria and eight of them had disease reactivation. The mortality rate was 52.3% and the mean time between diagnosis and death was 9.98 months (ranging from 0.5 to 46.9 months) Table 3.
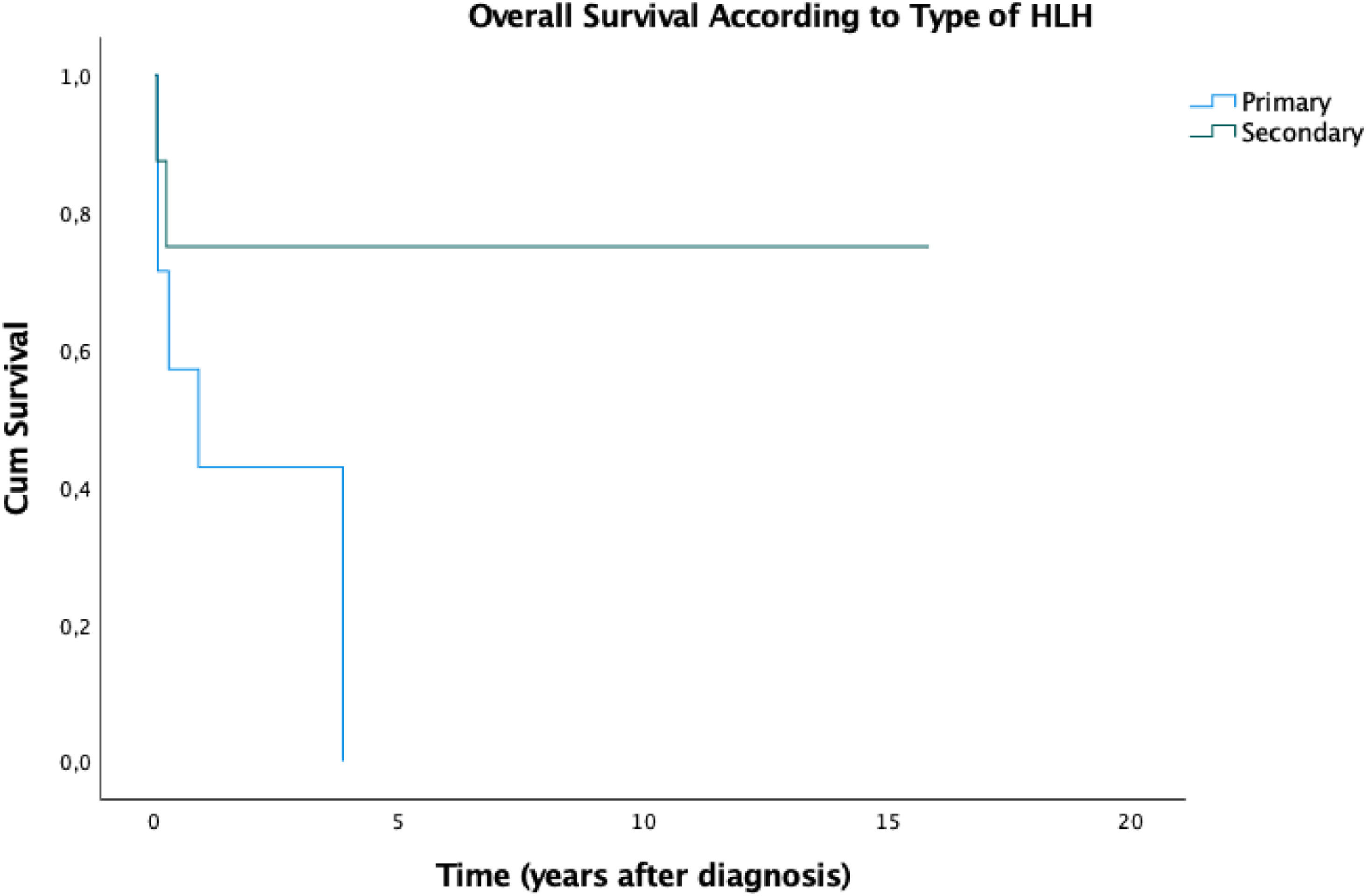
In our study of 21 patients, the estimated 5-year overall survival (OS) rate was 36.7% (Figure 1) and this OS remained until the end of the follow-up. We stratified the OS according to the type of HLH and the results are summarized in Figure 2. Patients with secondary disease had a better 5y-OS (75%), while no patients with primary disease were alive in five years. The percentage of living patients was 75% for primary disease, 28.6% for secondary disease and 16.7% for those who were not tested. Despite the better OS in patients with known secondary HLH, the results did not achieve statistical significance (p = 0.152). The difference in OS, considering the presence or absence of cytopenia, hypertriglyceridemia, hypofibrinogenemia, hemoglobin < 9 g/dl, neutrophils < 1000/ml and platelets < 100,000/ml was not statistically significant. An important result was the worst prognosis, associated with the reactivation of HLH. These patients demonstrated an estimated 5-year OS of 25% (p = 0.022), while all the patients who did not reactivate were alive until the end of the follow-up.
Discussion.")
.")
HLH is a consequence of a dysfunction in the immunologic system activation that can affect both children and adults. Recent data estimates an incidence of 1:300,000 newborns in North America.12 The actual incidence of HLH in Brazil is not well established, as the diagnosis is rarely suspected and difficult to confirm. After an exhaustive literature search aimed at HLH studies in the Brazilian population, we found a case series of seven patients treated at a referral institution between 2010 and 2012.15
Genetic HLH is the primary form affecting mainly children under the age of two, with a mean age of six months.8 It is inherited in an autosomal recessive, or X-linked pattern. The most frequent mutation associated with this form is located in the perforin gene - PRF1, resulting in a reduction of its expression. This protein is expressed in the bone marrow precursors and its uncontrolled activation results in a substantial discharge of inflammatory cytokines.9 The major immunological dysfunction observed in these patients is the severe impaired function of NK cells, due in part to the reduced number of NK cells.10 One third of our patients had the familial or immunodeficiency-associated forms and the molecular findings were compatible with the literature, including mutations of PFR1, UNC13D and STXBP2 genes. The reactive or secondary HLH is the acquired form and it can happen in all age groups, but it is most frequently observed in adults. Two thirds of our HLH cases were likely secondary, including six cases in which molecular testing was not available; the predominant trigger was infection-related, as described in the literature. Palazzi et al.11 demonstrated that 42–61% of this form of HLH originates from viral infections, among which Epstein-Barr virus (EBV), cytomegalovirus (CMV) and herpes simplex were recognized as the most prevalent.11 The pathogenicity is poorly understood, but a reduction in perforin and a deficiency of NK cells have been observed in these patients, suggesting that the primary infection may be responsible for a dysregulation in the inflammatory and immune response.10,11
In regard to the clinical presentation, our findings were similar to other series, with fever and hepatosplenomegaly as the most common symptoms. The HLH-94 study, one of the largest prospective cohort studies to date, analyzing 249 patients with HLH and able to identify that 97% of the patients presented with fever and 95% with organomegaly at diagnosis.13 These results are similar to those described by the International Registry of the Histiocyte Society, in which 122 cases were analyzed, identifying 97% of the patients as having splenomegaly at diagnosis.14
The median survival time of untreated patients with HLH is reported to be 2–6 months, 13 thereby the early diagnosis is crucial to the patient's prognosis. Once the etiology had been identified and patients classified according to the severity criteria, first-line therapy was implemented, following the HLH-94 and HLH-04 protocols. All of our patients started therapy less than 40 h from the suspected diagnosis.
Our 5-year survival rate was 36.7%. Ferreira et al.16 reported a mortality among patients in the study of 28.6% (2/7) and both deaths occurred in patients suspected of having primary HLH. In accordance with the literature, there was a survival advantage in patients with secondary HLH, in comparison to the primary form, 7,13,14 albeit not statistically significant, most likely due to the small number of patients (Table 4). On the other hand, in comparison, disease reactivation carried a statistically worse prognosis in our series.
ConclusionHLH is a rare disease with a poor prognosis, with a clinical presentation similar to other pathologies, which can delay the diagnosis. In our series, it was more common in females and children under six years of age. It is associated with a high mortality rate, especially in patients with disease reactivation and those with familial or immunodeficiency-associated forms, making genetic testing crucial for appropriate management and prompt SCT indication.


