

To document the experience of one referral service with patients diagnosed with Evans syndrome, the treatment and response and to briefly review current treatment strategies and results.
MethodsPatients enrolled in this study fulfilled criteria for Evans syndrome. Data were retrieved from the clinical files and electronic databases of the Department of Hematology, Hospital Universitario “Dr. José Eleuterio González”. Treatment modalities and response and the use of additional therapies were evaluated. The literature was reviewed in the context of the clinical course of the studied patients.
ResultsSix patients were diagnosed with Evans syndrome in the study period. Patient 1 was treated with steroids, relapsed twice and was again treated with steroids. Patient 2 treated initially with steroids plus intravenous immunoglobulin was subsequently lost to follow-up. A good response was achieved in Patients 3 and 4, who were treated with steroids plus rituximab; patient 4 also received danazol as a second-line therapy. However both relapsed and subsequently underwent splenectomy at ten and nine months, respectively. One patient, number 5, treated with steroids, danazol and rituximab did not relapse within four years of follow-up and Patient 6, who received steroids plus danazol did not relapse within three years of follow-up.
ConclusionEvans syndrome is an uncommon hematologic condition rarely diagnosed and not widely studied. Clinicians must have it in mind when evaluating a patient with a positive direct antiglobulin test, anemia and thrombocytopenia, since prognosis depends on its early recognition and opportune therapy, but even this leads to variable results.
Evans syndrome is a rare autoimmune disorder characterized by simultaneous or sequential presence of a positive anti-globulin test, autoimmune hemolytic anemia (AIHA) and immune thrombocytopenia (ITP).1 It is characterized by frequent exacerbations and remissions within a chronic course. Evans syndrome was first described in 19512 and it is recognized as a poor prognostic factor in autoimmune cytopenias.3 It is a rare disorder diagnosed in 0.8% to 3.7% of all AIHA or ITP cases.1 Its etiology and cause are unknown, but alterations in immune regulation mechanisms are documented.
This syndrome can be classified as primary or idiopathic when there is no associated disease, and secondary when it is associated with other autoimmune diseases, such as systemic lupus erythematosus (SLE), primary antiphospholipid syndrome, Sjögren syndrome, IgA deficiency, Hodgkin's disease and chronic lymphocytic leukemia.4 The diagnosis is made by exclusion of other pathologies, including infectious processes and malignant and autoimmune diseases. It presents with bicytopenia, which can coincide or occur separately or sequentially. After the appearance of the first cytopenia, the second may occur months to years later, which can delay diagnosis.5,6
Management of Evans syndrome remains a challenge. Response to treatment varies even within the same individual. Indications for treatment have not been established by evidence-based studies,5 in part due to the low frequency and heterogeneous nature of the disease. The first-line treatment for Evans syndrome is corticosteroids with or without intravenous immunoglobulin (IVIG).7 The range of options for second-line treatment includes immunosuppressive agents, the monoclonal antibody rituximab, chemotherapy or a combination of these agents. However, only a small percentage of patients achieve complete remission and these drugs have numerous side effects.8 Splenectomy may also be considered a second-line treatment. The majority of patients will respond to first or second-line therapy modalities, sometimes for several years. However, for patients with severe relapsing disease despite second-line therapy, other options have to be considered. The main third-line options are cyclophosphamide, alemtuzumab or stem cell transplantation.5
There is very limited information available in the literature regarding this infrequent syndrome; therefore we decided to present and discuss six cases diagnosed in our hospital over six years in order to call the attention of the physician to the importance of considering this disease when confronted with a patient exhibiting clinical and laboratory features compatible with Evans syndrome.
MethodsThis study was performed in accordance with the ethical standards of the Helsinki Declaration, including the provisions for patient informed consent. The Review and Ethics Committee of the institution approved the study.
The six patients included in this report were diagnosed between 2007 and 2012. All patients presented with AIHA and a positive direct antiglobulin test plus ITP. Clinical presentation included the usual features of hemolytic anemia: pallor, lethargy, jaundice, thrombocytopenia, petechiae, bruising and mucocutaneous bleeding.
There are no guidelines established for management of Evans syndrome, thus, for the purpose of this report, response was defined as resolution of all clinical symptoms and increase or no further decrease in both, platelet count and hemoglobin concentration. Relapse was considered to exist when patients presented with the same or similar clinical symptoms and laboratory data, including a positive direct antiglobulin test.
ResultsPatient characteristicsData of six patients, four women (66.64%) and two men (33.32%), fulfilling the diagnostic criteria of Evans Syndrome were retrieved from the clinical files and electronic databases (Table 1). Median age at diagnosis was 24 years. Both cytopenias occurred simultaneously in all cases. No cases of autoimmune neutropenia at diagnosis or during the clinical course were observed. Evans syndrome was considered idiopathic in one patient (16.6%) and was associated with one or more underlying diseases in the other five patients (83.4%; Table 1).
General characteristics of six patients diagnosed with Evans syndrome.
| Patient | Gender | Age | Date of diagnosis | Medical history before diagnosis |
|---|---|---|---|---|
| 1 | F | 10 | 2007 | Chronic ITP since age 4, treated with steroids |
| 2 | F | 14 | 2011 | SLE, ITP since age 10, treated with steroids |
| 3 | F | 42 | 2009 | SLE, AIHA |
| 4 | F | 19 | 2010 | SLE |
| 5 | M | 29 | 2011 | None |
| 6 | M | 29 | 2012 | SLE, APLA, PTT prolongued |
ITP: immune thrombocytopenia; SLE: systemic lupus erythematosus; AIHA: autoimmune hemolytic anemia; PTT: partial thromboplastin time; APLA: antiphospholipid antibody.
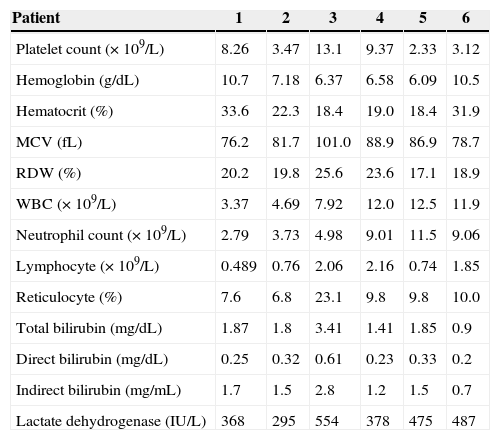
The complete blood count at diagnosis showed remarkable alterations (Table 2), including platelet counts ranging from 2.33 to 13.1×109/L (median: 5.8×109/L); hemoglobin concentration at presentation varied from 6.1 to 10.7g/dL (median: 6.9g/dL), the Mean corpuscular volume was within the normal range (76.2 to 101fL), but the red cell distribution width varied widely from 17.1 to 25.6% (median: 20%) reflecting the abundant presence of reticulocytes which ranged from 6.8 to 23.1% (median: 9.8%). Accordingly, indirect bilirubin concentration was increased in almost every case (median: 1.5mg/dL), and lactate dehydrogenase (LDH) values varied between 295 and 554U/L (median: 426.5U/L), reflecting the ongoing active hemolysis. The presence of anemia at variable degrees with hemolytic characteristics, including a high level of LDH and/or indirect bilirubin, with a repeatedly positive direct antiglobulin test and thrombocytopenia led to the diagnosis of Evans syndrome in all six patients.
Laboratory results at diagnosis of six patients with Evans syndrome.
| Patient | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| Platelet count (×109/L) | 8.26 | 3.47 | 13.1 | 9.37 | 2.33 | 3.12 |
| Hemoglobin (g/dL) | 10.7 | 7.18 | 6.37 | 6.58 | 6.09 | 10.5 |
| Hematocrit (%) | 33.6 | 22.3 | 18.4 | 19.0 | 18.4 | 31.9 |
| MCV (fL) | 76.2 | 81.7 | 101.0 | 88.9 | 86.9 | 78.7 |
| RDW (%) | 20.2 | 19.8 | 25.6 | 23.6 | 17.1 | 18.9 |
| WBC (×109/L) | 3.37 | 4.69 | 7.92 | 12.0 | 12.5 | 11.9 |
| Neutrophil count (×109/L) | 2.79 | 3.73 | 4.98 | 9.01 | 11.5 | 9.06 |
| Lymphocyte (×109/L) | 0.489 | 0.76 | 2.06 | 2.16 | 0.74 | 1.85 |
| Reticulocyte (%) | 7.6 | 6.8 | 23.1 | 9.8 | 9.8 | 10.0 |
| Total bilirubin (mg/dL) | 1.87 | 1.8 | 3.41 | 1.41 | 1.85 | 0.9 |
| Direct bilirubin (mg/dL) | 0.25 | 0.32 | 0.61 | 0.23 | 0.33 | 0.2 |
| Indirect bilirubin (mg/mL) | 1.7 | 1.5 | 2.8 | 1.2 | 1.5 | 0.7 |
| Lactate dehydrogenase (IU/L) | 368 | 295 | 554 | 378 | 475 | 487 |
MCV: mean corpuscular volume; RDW: red cell distribution width; WBC: white blood cell count.
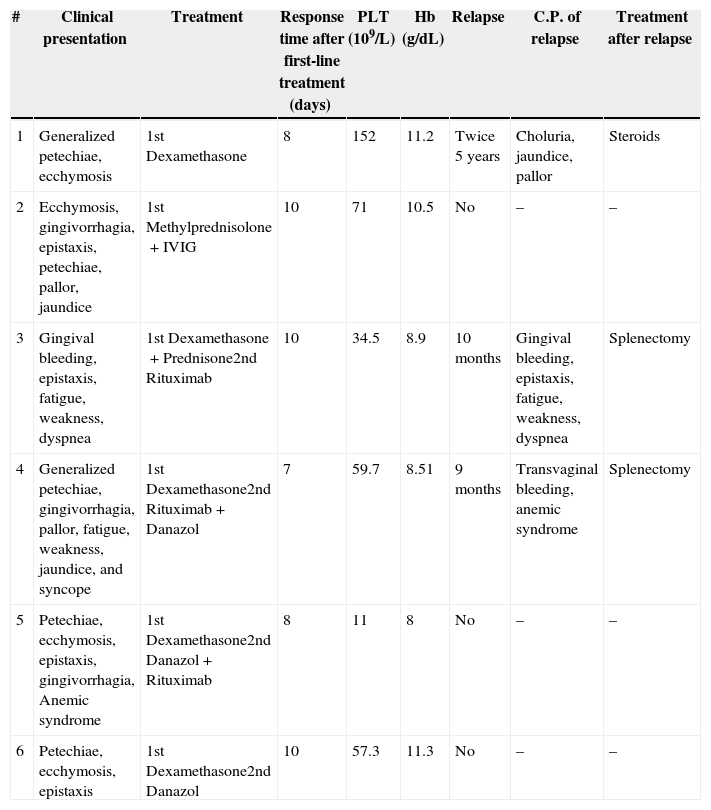
Detailed information regarding clinical presentation, treatment and evolution, as well as relapses and their therapy is shown in Table 3. Patient 1 was the only case in which steroids were successfully used as both first-line treatment and during relapses without additional medications; in all the remaining five cases a combination of therapies was needed to achieve response. Response times, as days needed for the increase in hemoglobin concentration and platelet count to take place, are shown in Table 3. Patients were discharged at a median of nine days (range: 3–12 days).
Clinical presentation, treatment, and relapse of six patients diagnosed with Evans syndrome.
| # | Clinical presentation | Treatment | Response time after first-line treatment (days) | PLT (109/L) | Hb (g/dL) | Relapse | C.P. of relapse | Treatment after relapse |
|---|---|---|---|---|---|---|---|---|
| 1 | Generalized petechiae, ecchymosis | 1st Dexamethasone | 8 | 152 | 11.2 | Twice 5 years | Choluria, jaundice, pallor | Steroids |
| 2 | Ecchymosis, gingivorrhagia, epistaxis, petechiae, pallor, jaundice | 1st Methylprednisolone+IVIG | 10 | 71 | 10.5 | No | – | – |
| 3 | Gingival bleeding, epistaxis, fatigue, weakness, dyspnea | 1st Dexamethasone+Prednisone2nd Rituximab | 10 | 34.5 | 8.9 | 10 months | Gingival bleeding, epistaxis, fatigue, weakness, dyspnea | Splenectomy |
| 4 | Generalized petechiae, gingivorrhagia, pallor, fatigue, weakness, jaundice, and syncope | 1st Dexamethasone2nd Rituximab+Danazol | 7 | 59.7 | 8.51 | 9 months | Transvaginal bleeding, anemic syndrome | Splenectomy |
| 5 | Petechiae, ecchymosis, epistaxis, gingivorrhagia, Anemic syndrome | 1st Dexamethasone2nd Danazol+Rituximab | 8 | 11 | 8 | No | – | – |
| 6 | Petechiae, ecchymosis, epistaxis | 1st Dexamethasone2nd Danazol | 10 | 57.3 | 11.3 | No | – | – |
PLT: platelet count; Hb: hemoglobin; C.P.: clinical presentation; IVIG: intravenous immunoglobulin.
Relapse was diagnosed when patients presented with same or similar symptoms as baseline and the simultaneous or sequential presence of a positive anti-globulin test, autoimmune hemolytic anemia and immune thrombocytopenia. An appropriate follow-up was documented in five of the six patients. Three patients relapsed within nine months to five years.
One patient (Patient 2) responded satisfactorily to initial steroid therapy and was subsequently lost to follow-up. Two patients, 5 and 6, had not relapsed 45 and 32 months after the initial diagnosis, respectively. Patients 1, 3, and 4 relapsed. Patient 1 presented two relapses, the first one, five years after diagnosis, was treated with steroids, obtaining complete remission until a year later when she presented moderate thrombocytopenia and severe AIHA. This episode was again treated as in her first relapse, with steroids, obtaining a third remission lasting 24 months to date. Patient 3 had a relapse at ten months after diagnosis; she was treated with dexamethasone, prednisone and rituximab, with no response; splenectomy was performed at this time. This patient has not relapsed to date, 57 months after splenectomy. Patient 4 relapsed at nine months. She was treated with splenectomy and has not suffered further relapse 32 months after removal of the spleen; clinical course and therapy for these five patients are summarized in Table 3.
DiscussionEvans syndrome is a rare autoimmune regulation disorder whose exact pathophysiology is unknown. Clinically, the disease consists of simultaneous or sequential autoimmune hemolytic anemia and immune thrombocytopenia, with or without bleeding from mucous membranes and petechiae, and/or immune neutropenia in the absence of any other cause.1
There is a decrease in serum immunoglobulins, especially IgG, IgM and IgA.9 Savasan et al.4 described evidence of lymphoid hyperplasia and hyperactivity with deregulation of the APO-1 antigen, which is expressed on activated T and B cells, and is in intimate relationship with the immune pathway inducing apoptosis. Wang et al.9 showed proportions of diminished T4 and increased T8 cells with a decreased T4:T8 index. Decreases in the production of interleukin-10 and gamma-interferon have been reported, and it is postulated that this causes the activation of B cells producing auto-antibodies. These alterations are not exclusive to Evans syndrome as they can be found in other immune diseases, thus it remains to be established if they are indeed a cause of the disease or merely associated immune phenomena.
The differential diagnoses for Evans syndrome include thrombotic thrombocytopenic purpura, chronic cold agglutinin disease, other causes of acquired or hereditary hemolytic anemia, and drug-induced hemolytic anemia and/or thrombocytopenia. Its exact incidence remains unknown. In a review of adult patients with immunocytopenias including 766 patients with 399 cases of AIHA and 367 cases of thrombocytopenia, Evans syndrome was diagnosed in only six (0.78%) patients.10 The largest reported series of Evans syndrome in pediatric patients included 164 cases of ITP and 15 of AIHA; only seven (4.1%) children were diagnosed with the syndrome.11
Evans syndrome can be grouped into primary or idiopathic, when there is no associated autoimmune disease and secondary when it is associated with another autoimmune disease, such as SLE, Sjögren syndrome, Hodgkin's disease, or chronic lymphocytic leukemia, among others.4 In adults, an underlying cause can be expected in 70% of the cases.12 In our study five of six patients had an autoimmune disease diagnosed before they developed Evans syndrome.
The disease is characterized by frequent exacerbations and remissions within a chronic course and response to treatment varies even within the same individual.5 Most patients require treatment although occasional spontaneous remissions have been reported, as was the case in one of 42 patients.13 Indications for treatment have not been established by other evidence-based studies. However, it is reasonable and usual to treat symptomatic patients with low blood counts; not all asymptomatic patients with low counts require treatment and the decision to treat or not should be considered according to each individual case.5 Most of the data are anecdotal and inconclusive with difficult interpretation because of the concomitant use of corticosteroids and other treatment modalities.13,14
Corticosteroids remain the mainstay of therapy for control of the acute symptomatic cytopenias, with good initial results, despite lack of controlled trials demonstrating their efficacy.5 Pui et al., on describing the clinical features and long-term follow-up of seven children with Evans syndrome, found that in all six children requiring treatment, prednisolone at a daily dose of 1–2mg/kg resulted in remission; however, this response was lost upon dose reduction and/or during acute viral infections.11 In the current study, all six patients were treated with at least one corticosteroid. The initial dose of methylprednisolone (1.0g IV diluted in 100mL, every 24h for four doses) was used only for Patient 2. The treatment strategy for Patient 3 was prednisone administered at a dose of 40mg p.o. every 12h until reaching a response or for four weeks. When using dexamethasone, as was the case in five of the six patients in this study, the dose was 40mg IV every 24h for four doses, with this obtaining a good clinical response in all cases, with improvement in platelet count and hemoglobin concentration but without reaching normalization.
If steroids are ineffective or unacceptably high doses are required to maintain remission or if toxicity occurs, the most commonly used treatment is IVIG. Various doses have been suggested, most commonly 0.4g/kg/day for four doses with some other authors recommending higher doses (up to 5.0g/kg) to improve response in AIHA.15 In our patients, only one received IVIG as part of her initial treatment (Patient 2) at a single dose of 1.0g/kg with good response.
Second-line treatment includes immunosuppressive agents (cyclosporine and mycophenolate mofetil), chemotherapy (vincristine and cyclophosphamide), danazol and monoclonal antibodies (rituximab and alemtuzumab).5 Although splenectomy has traditionally been used as initial second-line therapy in patients with autoimmune cytopenia (ITP or AIHA) who did not respond to or relapsed after standard therapy with steroids with or without IVIG, the role of splenectomy in the treatment of Evans syndrome is not clearly established.5 Overall, the response rate to splenectomy is lower than the 70–75% reported in chronic ITP. There are few data for Evans syndrome; thus accurate response rates cannot be cited.16 Splenectomy often produces immediate improvement or even complete normalization of blood counts. This response is often transient and relapse occurs in most cases 1–2 months post-splenectomy independently of whether steroids are continued postoperatively.11,13,14 However, splenectomy occasionally results in sustained remission (one of eight patients remained in complete remission at six years post-splenectomy in one report),15 and there is some evidence that splenectomy may be beneficial in reducing the frequency of relapses and lowering the maintenance dose of steroids.11,13,14 Only two patients in our group were splenectomized (Patients 3 and 4) due to relapse.
Danazol was administrated in three out of six patients in combination with steroids. These three individuals had good initial responses; however, two relapsed and required additional treatment with one being successfully splenectomized (Patient 4) after the first relapse. On the other hand, rituximab at a dose of 375mg/m2 was used in three of six patients, with no response in two patients (Patients 3 and 4), leading to splenectomy. Only one patient (Patient 5) responded satisfactorily to rituximab.
The majority of patients will respond to first or second-line therapy, and this response can last several years. However, for patients with severe, relapsing disease despite second-line therapy, other options have to be considered. The main third-line options are cyclophosphamide, alemtuzumab or stem cell transplantation.5 Three of the patients in this study relapsed within nine months to five years, however none of them required a third-line treatment. This wide variability and unpredictability in relapsing times have been reported previously.13,14
There are important issues to define with respect to treatment and response criteria for Evans syndrome. In this regard it is not clear whether it is important for steroids to be administered at the same time as IVIG, although this is common practice. The majority of patients, unfortunately, relapse as therapy is tailed off and second-line therapy will be required. The choice of which second-line agent to use depends upon clinical criteria, particularly the age of the patient, severity of the disease and its natural history because all of these treatments have significant short- and long-term side effects.5 It is pertinent to underscore that there are no standardized response criteria for Evans syndrome, in contrast to ITP where an increase in the platelet count to ≥30.0×109/L is considered as response (R) and ≥100.0×109/L on at least two separate occasions at least seven days apart as a complete response.17
Our six patients presented with severe clinical manifestations and received steroids with or without IVIG, danazol or rituximab in order to prevent or delay complications that can endanger life.18 After failure of rituximab, splenectomy was performed in two of three relapsing patients, underscoring the importance of this intervention in the management of the disease, and suggesting that, at least in the setting of autoimmune associated Evans syndrome, the addition of rituximab does not guarantee therapeutic success.
For an early diagnosis and successful treatment, Evans syndrome must be kept in mind whenever hemolytic anemia with a positive direct antiglobulin test and thrombocytopenia are present, especially in patients with an autoimmune disease.
Conflicts of interestThe authors declare no conflicts of interest.
The authors gratefully acknowledge Sergio Lozano-Rodriguez, M.D. for his review of the article.


