

The Evans syndrome (ES) is a rare, often chronic, relapsing and treatment-refractory hematological disorder. We described the clinical features, diagnostic workup, treatment and outcome in patients with ES.
MethodWe performed a retrospective chart review of patients aged < 18 years with ES admitted to a tertiary center in Brazil from 2001 to 2021. The analysis of the data was primarily descriptive, using median, interquartile range and categorical variables presented in absolute frequencies.
Main resultsTwenty patients (12 female, 8 male) were evaluated in this study. The median age at the initial cytopenia was 4.98 years (1.30–12.57). The ES was secondary in nine cases (45%), of which six patients (30%) showed autoimmune disease (AID) or primary immunodeficiencies (PID) and one presented a spontaneous recovery. Steroids and intravenous immunoglobulin were first-line therapy in 19 cases. Twelve patients (63%) required second-line treatments (rituximab, cyclosporine, splenectomy, sirolimus, cyclophosphamide, mycophenolate mofetil, azathioprine and eltrombopag). The median follow-up period was 2.41 years (1.4 –7.52). One patient (5%) died of underlying neuroblastoma, one case (5%) was lost to follow-up and four patients (20%) received a medical discharge. The median age for the 14 remaining cases was 12.6 years. Twelve patients (85.7%) were in complete response (CR) with no therapies. Two patients (14.3%) were in CR with chronic therapy.
ConclusionAs ES may be a symptom of AID and PID, a thorough rheumatological, immunologic and genetic workup and a careful follow-up are essential. The second-line treatment remains a dilemma. Further prospective studies are needed to address the optimal therapeutic combinations, morbidity and mortality in this disorder.
The Evans Syndrome (ES) or Evans Fisher was first described by Robert Evans and associates in 1951 as the association of Autoimmune Hemolytic Anemia (AIHA) and Idiopathic Thrombocytopenic Purpura (ITP) [1]
From the 80s, the association of AIHA and ITP started to be questioned and since 2000, some researchers admitted ES as the presence of AIHA associated with immune thrombocytopenia (IT) and/or immune neutropenia (IN), with the involvement of platelets and/or neutrophils possibly occurring simultaneously or sequentially [2,3]. It is rare in pediatrics and the incidence in children under 13 years of age was 0.5 and 1.2 / 1,000,000 in 1981 and 2015, respectively. The prevalence ranged between 6.7 and 19.3 / 1,000,000 person-years [3].
The course of ES is characterized by a heterogeneous chronic disease with clinical variability at onset, spontaneous remissions and exacerbations. Clinical features are pallor, weakness, fatigue, jaundice, petechiae, ecchymosis, gingivorrhagia and epistaxis [3,4]. Hepatomegaly, splenomegaly and lymphadenopathies can occur [4].
The ES is classified as Primary (diagnosis of exclusion) or Secondary (associated with hematological malignancies, systemic autoimmune disorders, primary immunodeficiencies, chronic infections and post-transplant) [3,5]. The determination of the primary or secondary nature of the ES is important because this can interfere with its management and prognosis.
This article describes the clinical features, diagnostic workup, treatment and outcomes in children and adolescents diagnosed with ES at our institution over the past 20 years.
MethodsAfter the Institutional Review Board approval (47746721.5.0000.0068) and written informed consent from parents and eligible patients, we performed a retrospective chart review of all pediatric patients admitted with ES seen at a Tertiary Public Hospital in São Paulo, Brazil from 2001 to 2021.
We searched the electronic medical records for patients under 18 years of age with a diagnosis of AIHA (International Classification of Diseases version 10, codes D59.1 and D59) and ITP (code D69.3).
All patients with the diagnosis of ES fulfilling the eligibility criteria were included: the occurrence simultaneously (up to one month) [6] or sequentially (without clearly defined periods) of AIHA and/or IT and/or IN. The AIHA was defined as anemia (Hb < −2SDS) with features of hemolysis (low haptoglobin level and/or elevated lactate dehydrogenase and/or unconjugated hyperbilirubinemia and/or reticulocytosis) and a positive direct antiglobulin test (DAT). The IT was defined as a platelet count less than 100 × 109 /L, in the absence of other causes or disorders that may be associated with thrombocytopenia [7]. In case of concomitant active AIHA, the presence of mild splenomegaly was not an exclusion criterion for IT. The IN was defined as an absolute neutrophil count below 1.5 × 109/L and 1.0 × 109/L in white and brown/black participants, respectively, with or without anti-neutrophil autoantibodies, lasting more than six months without infectious or drug-induced myelotoxicity [6,8]. Patients were included on the first date on which the AIHA and/or IT and/or IN had been recorded.
The AIHA and IT were defined as severe if the Hb level < 7g/dL and platelet count < 10 × 109/L, respectively [6].
The exclusion criteria were: patients with isolated DAT without anemia; presence of schizocytes on the blood smear with a negative DAT, suggesting thrombotic microangiopathies and hereditary hemolytic anemia.
The primary and Secondary ES were analyzed. In primary ES, no hematological malignancies, autoimmune disease (AID), chronic infections, or primary immunodeficiencies (PID) could be identified during the entire follow-up.
The secondary ES was associated with PID genetically identified by exome sequencing, systemic lupus erythematosus (SLE), based on the American College of Rheumatology (ACR) classification criteria (2019) [9], and chronic infections (Epstein-Barr, human immunodeficiency virus, Helicobacter pylori, Cytomegalovirus and Hepatitis C virus) [5].
Additional secondary causes were also included, for example, post-transplant ES cases following unrelated bone marrow transplantation or solid organ transplantation and drug-induced hemolytic anemia and/or thrombocytopenia [5].
To assess treatment efficacy, a complete response (CR) was defined by the normal hemoglobin level range for age, in the absence of any transfusion without features of hemolysis and platelet count > 100 × 109 /L. Patients who showed hemoglobin < 8 g/dL and platelets < 30 × 109 /L despite treatment were considered to show no response (NR). Partial response (PR) was defined by hemoglobin and platelet levels between CR and NR.
The data analysis was primarily descriptive. Continuous variables were reported as median and interquartile range (1st to 3rd quartile). Categorical variables were presented in absolute frequencies.
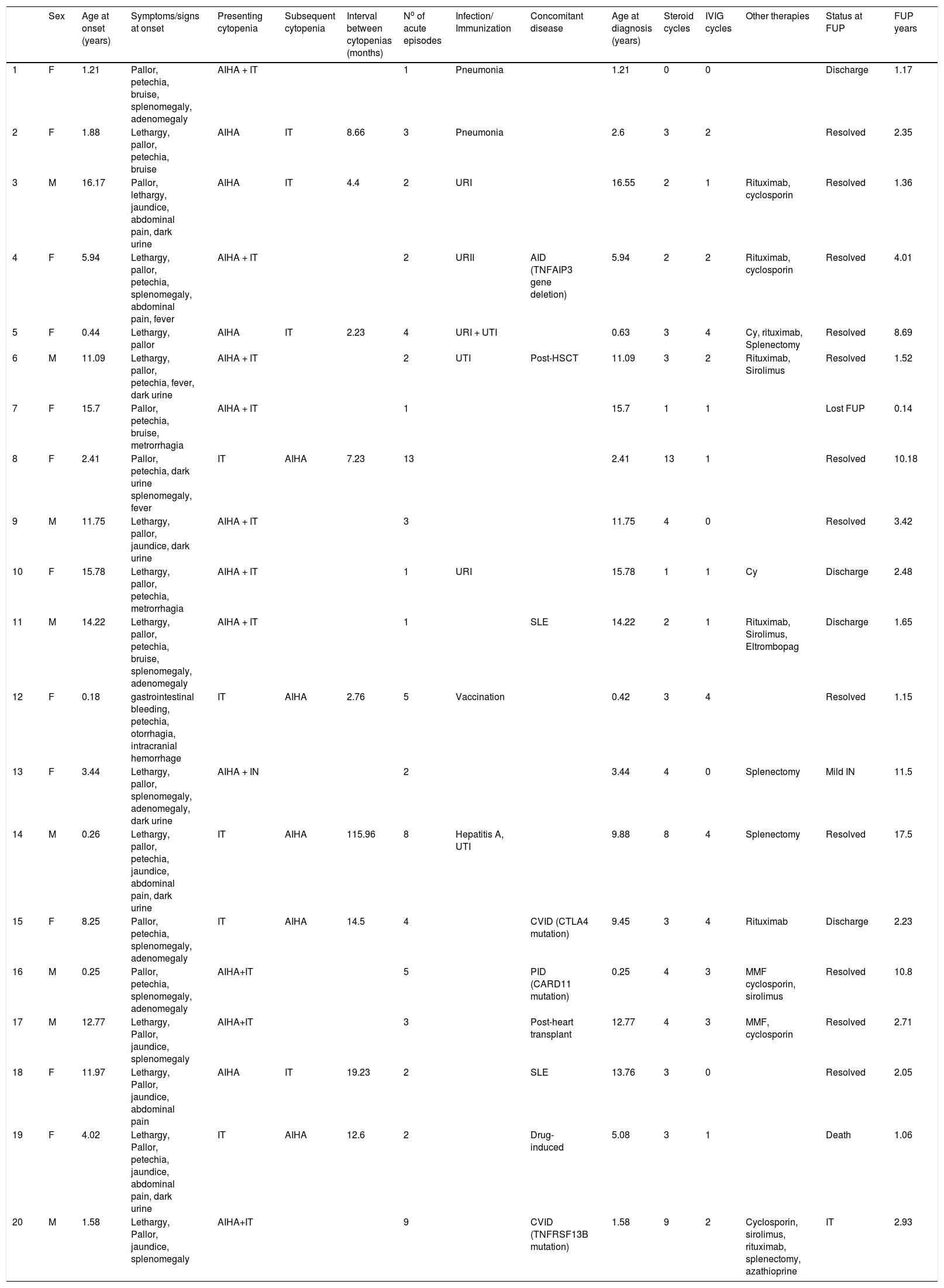
ResultsTwenty patients with ES were evaluated in this study. The sex ratio was 1.5 (12F/ 8M). The median age at initial cytopenia was 4.98 years (1.30–12.57), whereas the median age at diagnosis was 7.69 years (1.78–13.51). The patient clinical and laboratory characteristics are presented in Tables 1 and figure 1.
Clinical characteristics of pediatric patients with ES.
. IVIg, intravenous immunoglobulin.")
Both cytopenias occurred simultaneously in 11 cases (55%). The IT preceded the onset of AIHA in 5 cases (25%), whereas AIHA was the first manifestation in 4 patients (20%). A concurrent IN was observed in 1 patient (5%) at the time of diagnosis of the ES. When the cytopenias occurred sequentially, the median delay between both cytopenias was 8.66 months (3.17–18.04). The patients experienced a median range of 2.5 exacerbation episodes (2–4.75) and the median time between acute events was 6.62 months (1.58–8.97). The hematological presentation was severe in 14/20 (70%) AIHA patients and in 7/20 (35%) IT patients. In the neutropenic patient, clinical consequences and anti-neutrophil autoantibodies were not identified. The granulocyte colony-stimulating factor was not used for prophylaxis.
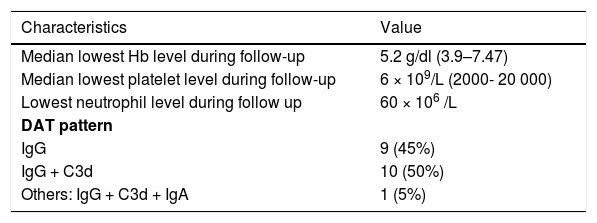
The DAT type was consistent with the IgG plus complement (C3d) in ten patients (50%) or IgG in nine cases (45%). One patient (5%) presented the DAT type IgG plus C3d and immunoglobulin A (IgA). (Table 2)
Fatigue or lethargy (70%), jaundice (35%), and hemoglobinuria (35%) were the most commonly presented symptoms. Nine patients (45%) presented hepatosplenomegaly or lymphadenopathy. Mucocutaneous bleeding was reported in 13 patients (65%) and severe bleeding (intracranial hemorrhage) occurred in one case (5%).
Until the last clinic visit, the ES was considered primary in eleven patients (55%), as three of them were under screening for PID and ES was secondary in nine cases (45%): PID (n = 3), AID (n = 3), post-hematopoietic stem cell transplantation (HSCT, n = 1), post-heart transplant (n = 1) and drug-induced (n = 1) (Table 1).
Eight (40%) patients had acute infectious disease at the time of the first cytopenia (pneumonia (n = 2); upper respiratory tract infection (n = 3); urinary tract infection (n = 2); concomitant hepatitis A and urinary tract infection (n = 1)). One patient (5%) received the diphtheria, tetanus and pertussis (DTP) vaccine, plus pneumococcal and polio vaccines, 15 days before the cytopenia onset.
One patient presented spontaneous hematological recovery within 1.4 months of diagnosis.
Nineteen patients received at least one course of steroids. Three patients (15.7%) received monotherapy (methylprednisolone 30 mg/kg/d × 3 days, followed by oral prednisone 1-2 mg/kg/d, for at least 21 days). Sixteen cases (84.2%) were treated with steroids and intravenous immunoglobulin (IVIg: 1-2 g/kg for two days). Nine patients (47.3 %) showed response to first-line therapies (6 CR and 3 PR). The CR occurred in a median recovery time of 13.5 days (2–21.75) from the first cytopenia. Ten patients (52.6%) showed no response and one of them died of an underlying cancer.
Second-line treatments were required in 63% of the patients (9 NR and 3 PR). The main second-line therapies were: rituximab (n = 7) with a complete response rate of 71.5% (n = 5); cyclosporine (n = 5); splenectomy (n = 4), sirolimus (n = 4); cyclophosphamide (n = 2), mycophenolate mofetil (n = 2); azathioprine (n = 1), and; eltrombopag (n = 1).
The median follow-up period (FUP) from the first cytopenia was 2.41 years (1.4–7.52). Overall, one patient (5%) died at age of five years old of underlying neuroblastoma; one patient (5%) was lost to follow-up, and; in 4 cases (20%), the follow-up was discontinued because the patient was considered cured.
At the time of the last follow-up, the median age for the 14 remaining patients was 12.6 years (7.99-15.25). Twelve cases (85.7%) were in CR with no therapies. Two patients (14.3%) were in CR with chronic therapy, with one of them receiving sirolimus (patient 16) and the other one using azathioprine plus prednisone post-splenectomy (patient 20).
No patient developed infectious complications presumed to be secondary to immunosuppression.
DiscussionWe reported on the largest contemporary single institutional series of pediatric ES study in Latin American.
Most of our patients were females (1.5: 1 ratio). This is in contrast with the unexplained tendency for the male predominance in younger patients and female predominance in older ones [3,6,10].
In contrast to the French national pediatric network, the largest prospective observational study in pediatric ES, that reported 25% of the cases associated with genetic familial predisposition, none of our patients had a known similar condition [6]. No specific ethnic predilection is noted among patients with ES [11].
According to the literature, we found simultaneous AIHA and IT at the initial presentation in 55% of the cases, IT preceded the onset of AIHA in 25% and AIHA was the first manifestation in 20% [6,12]. In our study, 5% of the children presented AIHA and IN, lower in comparison to the 20-24% reported in the French, North American and Canadian databases [6,13]. We found that the median time of sequential autoimmune cytopenias was 0.72 years (0.26–1.5) following the diagnosis from the first autoimmune cytopenia, differing from other studies which reported an average of 2.4– 2.9 years [6,12].
The severeness and outcomes of anemia and thrombocytopenia can be different in a single patient. The severe hematological presenting values observed in our patients were higher for AIHA (70%), compared to 49% reported in the French cohort. Severe IT was identified in 35% of our cases, in contrast with 51% in the French study [6].
Our patients experienced a median of two acute episodes and a median time interval of 6.62 months between exacerbations, lower compared with four episodes (1–7) and 15 months (4–96), respectively, reported in Italy [8]. The mortality rate was 5% (FUP 2.41 years), lower than the 10 % (FUP 6.5 years) rate reported in France [6,12]. These different results may be related to our reduced sample size and inferior follow-up time. Although mortality is not directly associated with cytopenia complications in all cases, it is notable that the ES mortality and morbidity are higher than in any other autoimmune cytopenia [12].
The ES reflects a state of immune dysregulation with multiple autoimmune cytopenias. In clinical practice, cases of ES may show or precede a variety of underlying diseases or conditions which may influence the management and outcome. The commonly identified diseases driving ES are PID, most often common variable immunodeficiency disorder (CVID) [14,15,16], AID, most often SLE [6], and autoimmune lymphoproliferative syndrome (ALPS) [17,18].
The AID and PID account for approximately 56.5% of all cases [14]. According to the literature, AID and PID are observed in 30.4% and 26.1%, respectively [14]. In our study, six patients (30%) showed these secondary etiologies (AID 15%, PID 15%).
The ES was secondary to CVID (CTLA4 gene mutation, TNFRSF13B gene mutation) in two patients (10%) and associated with the CARD11 gene mutation in one patient. Of note, as ES may be a presenting symptom of PID, it is essential to guarantee a thorough immunologic and genetic workup at the clinical onset and a careful follow-up [15,19].
Although two of our patients (10%) presented ES (mean time 1.42 years) before SLE, ES is one of the rare presentations in SLE. According to a multicenter Brazilian SLE cohort study, ES, preceding the diagnosis, was identified in 1.3% of this population and these cases are characterized by the lack of the typical signs and symptoms of lupus [20].
None of our patients were diagnosed with ALPS, in contrast to the literature data, which reports a prevalence of 30–47% of ES cases [14,18].
According to the Italian Association of Pediatric Hematology and Oncology HSCT Registry, autoimmune hematological disease (AHD) is a relatively rare complication of the HSCT course. This study reported AHD in 2.1% of the children who underwent allogeneic HSCT at nine Italian centers and ES, as a manifestation of AHD, was diagnosed in 15% of the children at a median of 9.2 months after HSCT [21]. In our study, one patient (5%) affected by adrenoleukodystrophy, developed ES six months after HSCT. Nonmalignant underlying disease, transplantation from alternative donors and cord blood as a source of HSCT represented risk factors statistically associated with AHD. The frequency of AHD observed in transplanted children could be explained by the more competent immune system before HSCT in nonmalignant disease, the immunological dysregulation due to the use of calcineurin inhibitors or antithymocyte globulin administered before unrelated donor HSCT, viral infections, graft-versus-host disease and delayed immune reconstitution [21,22].
One of our cases (5%) was diagnosed with secondary ES 5.3 years after heart transplantation. A retrospective single center American study reported autoimmune cytopenia (AIC) in thirteen patients (6.9%) out of 188 heart transplant pediatric patients. The median time from the transplant to the first immune cytopenia in this study was 3.6 (0.7-4) years. The ES was common and occurred in 61.5% of the cases. These have been shown to be related to a range of factors, including chronic immunosuppression, especially tacrolimus, blood antigen incompatibility and infections. Additionally, heart transplantation may lead to a thymic dysfunction due to partial or total thymectomy, with an impact on immune modulation, which may consequently influence the pathogenesis of AIC [23].
In contrast to the French national pediatric network, which reported 10% of cases associated with chronic infections, none of our patients had a similar condition [6].
One patient (5%) had a history of preceding medications. The positive DAT, with a negative eluate test, was consistent with drug-induced hemolytic anemia. No specific test for the detection of drug antibodies was performed. The estimated incidence of AIHA is approximately 1 per million/year. More than 130 drugs have already been described as potential triggers. The most common ones include second and third generation cephalosporins, diclofenac, rifampicin and fludarabine. The clinical onset may occur within hours to months of exposure to the drug. The management includes discontinuing the suspected medicine and improvement is expected to occur in 1-2 weeks [24]. In our study, as the child was critically ill (metastatic neuroblastoma), it was difficult to identify which drug-induced ES it was. No response to steroids and intravenous immunoglobulin was observed. There was no clinical status to withdraw possible associated medications and the patient died due to cancer complications 1.2 years after the first cytopenia.
Moreover, no hematological malignancies were observed, which is opposed to the Danish nationwide study that described hematological malignancy-related ES in 19% of the ES cases [10].
Management of patients with ES is a challenge. Although some patients do not require treatment due to the transient and mild nature of their cytopenia, the clinical management and treatment of moderate or severe forms have no established guidelines or current evidence in the literature [25,26].
The frontline treatment is steroids, especially in cases in which AIHA is clinically dominant. The IVIG can be preferred in cases with pronounced thrombocytopenia, when steroids are ineffective or unacceptably high doses are required to maintain remission, or if toxicity occurs [10,26]. In our study, six patients (31.6) had CR following first-line therapies (steroid: 15.8%; steroid plus IVIG: 15.8%), lower than in the literature (steroids: 80–85%; IVIG 60-87%) [25,26]. The resistance to initial therapy in our patients may be related to their high prevalence of secondary ES. In these cases, studies have shown a higher demand for second-line treatments compared to the primary etiology [6,8].
Second-line therapies are used when patients develop treatment failure to first-line drug regimens (after three weeks) or experience a relapsing cytopenia, steroid dependency, or a chronic disease [25]. These treatments include monoclonal antibodies (rituximab and alemtuzumab), chemotherapy (cyclophosphamide), immunosuppressive agents (sirolimus, cyclosporine and mycophenolate mofetil), thrombopoietin receptor agonists (TPO-RAs), azathioprine and bortezomib [25]. The choice of the appropriate treatment should take into consideration the immunological background of the disease and the severity of clinical symptoms [25].
The management with Rituximab (375 mg/m2/week for four consecutive weeks) was the most common second therapy and our response rate was close to 76%, described in previous publications [4]. Despite the lack of prospective trials, this monoclonal antibody is currently preferred to splenectomy due to the lower risk of infective complications. The expected time to response is 3–6 weeks and up to 12 weeks must be considered [22] A long-lasting response in a mean follow-up of 2.4 years has been described in 65% of the patients. The main side-effect concern includes prolonged hypogammaglobulinemia which is a particular risk in patients with known underlying immunodeficiency [22,27].
In addition to rituximab, we administered cyclosporine (n = 5), sirolimus (n = 4), mycophenolate mofetil (n = 2), cyclophosphamide (n = 2) and azathioprine (n = 1). Cyclosporine was indicated as a corticoid-sparing or maintenance agent. Its use must be considered in severe refractory conditions, especially in the context of multidrug treatment [4]. Sirolimus is an effective and safe option in highly refractory ES, with a particularly excellent response in ALPS, and two of our cases who received sirolimus had a CR [16]. Mycophenolate mofetil (MMF) induced the CR in one patient and according to the literature, it has shown a sustainable response rate of 80% [4]. Cyclophosphamide induced CR in two patients. This alternative treatment was based on one case report, in which a combined immunosuppressive therapy including cyclophosphamide was successfully used in pediatric ES [28]. One patient, with no response to steroids, IVIG, monoclonal antibodies, immunosuppressive agents and splenectomy, received azathioprine associated with steroids. The patient presented a CR with this combined therapy. Although azathioprine has been shown to be able to control the disease in adult patients (response rate: 45-71%) [3], there are no similar data concerning the use of this drug for pediatric ES.
Eltrombopag (TPO-RA) is an option for second-line treatment in cases of ES with severe thrombocytopenia, refractory to steroids, splenectomy, IVIG and rituximab. Our patient (n = 1), without response to systemic immunomodulation (rituximab plus sirolimus), showed the recovery of the platelet count after the start of the eltrombopag treatment. According to the literature, the expected efficacy rate with this drug is 82% [4,25].
A splenectomy was performed in four (25%) patients. However, the procedure did not avoid the occurrence of the second episode. All patients had a relapse at a median time of 19.7 months after the procedure. A splenectomy should be considered only after the failure of other alternative drugs available for refractory patients and should not be performed in patients with ALPS [4,25]. Despite the limited data, response rates for splenectomy in ES are considerably heterogeneous, between 0% and 66% [4].
Finally, although hematopoietic stem cell transplantation (HSCT) may represent the possibility of definitive treatment for ES, scarce reports have been documented to guide treatment decisions. Multicenter research trials are necessary to further address this issue [4].
Our analysis has several limitations related to its retrospective design, including small sample size and single-center involvement. As ES is an uncommon condition and most published cohorts are small, it is difficult to compare our results with other studies and drawing clinical conclusions from limited data can be challenging.
In conclusion, ES is a rare, often chronic, relapsing and treatment-refractory hematological disorder in the pediatric population, in which mortality and morbidity are higher than in any other autoimmune cytopenia. This review of 20 cases gives a detailed description of the heterogeneity of the initial manifestation and highlights the different etiologies. We showed that most cases were considered secondary and associated with AID and PID. The knowledge that ES may be a presenting symptom of AID and PID is essential to guarantee an extensive thorough rheumatological, immunologic and genetic workup and a careful follow-up. Most of the patients received a second-line treatment, for which the best choice remains a dilemma. A multicenter and prospective trial with an adequate number of cases is essential to address the optimal therapeutics combinations, efficacy, morbidity and mortality in this disorder.



