
Intravascular large B-cell lymphoma (IVLBCL) is a rare subtype of extranodal large B cell lymphoma, characterized by the infiltration and growth of the neoplastic cells within blood vessel lumen, excluding larger arteries and veins.1,2
It typically presents with wide dissemination in extranodal sites, with a preference for the bone marrow but any organ can be affected. Nodal involvement is not usually seen. The clinical course of the disease is aggressive, and outcomes are dismal.
Two main variants have been recognized, with differences in clinical presentation that differ according to the geographical origin of the patients3,4; a so-called classic variant mostly seen in western countries, characterized by clinical symptoms related to the organs involved, with high frequency of Central Nervous System (CNS) and skin involvement3 and an hemophagocytic-syndrome like variant preferentially seen in Asia showing hemophagocytic syndrome, hepatomegaly, bone marrow involvement and fever.5 Despite the fact that morphologic examination is a key tool for the diagnosis of hemophagocytosis, it´s been recognized that hemophagocytosis-associated IVLBCL frequently lack this typical morphologic finding and this variant´s diagnosis grounds on the presence of a combination of clinical and laboratory features described above.2 Owing to the rarity of this entity, and the high prevalence of non-specific symptoms at presentation such as fever, general fatigue, loss of appetite without lymphadenopathy, diagnosis is often delayed, compromising patient´s outcomes.
Due to the infrequency of this lymphoma there is no standard of care treatment and anthracycline-based chemoimmunotherapy is recommended as in diffuse large B cell lymphoma NOS (DLBCL) and CNS study and prophylaxis of relapses at this site is advised. The role of autologous stem cell transplantation after first line treatment is discussed, but some experts advocate its use in young and fit patients.
Case presentationA 61-year-old male with no comorbidities was admitted to hospital due to daily fever over a month with no evidence of an infectious origin. Upon admission he was febrile, without any other relevant findings on physical examination.
Cultures were negative as well as viral and serological tests. Specific microorganisms were searched for with negative results, transesophageal echocardiogram and PET/TC scan were normal.
Fever persisted despite wide spectrum antibiotics empirically administered and the patient´s general condition worsened, with development of multiorgan disfunction syndrome (Hemoglobin 8.2 gr/dl, 59,000/mm3 Platelets; aPTT 50s, prothrombin time 40% plus liver and renal disfunction) Ferritin level > 10,000 ng/mL, LDH > 800 mUI/mL and hypertrigliceridemia were observed which led to the suspicion of hemophagocytic lymphohistiocytosis.
Bone marrow aspirate and biopsy were performed. On the aspirate no clear evidence of hemophagocytosis was found, however, IL-2 soluble receptor (sIL-2r) was 17,000 U/ml.
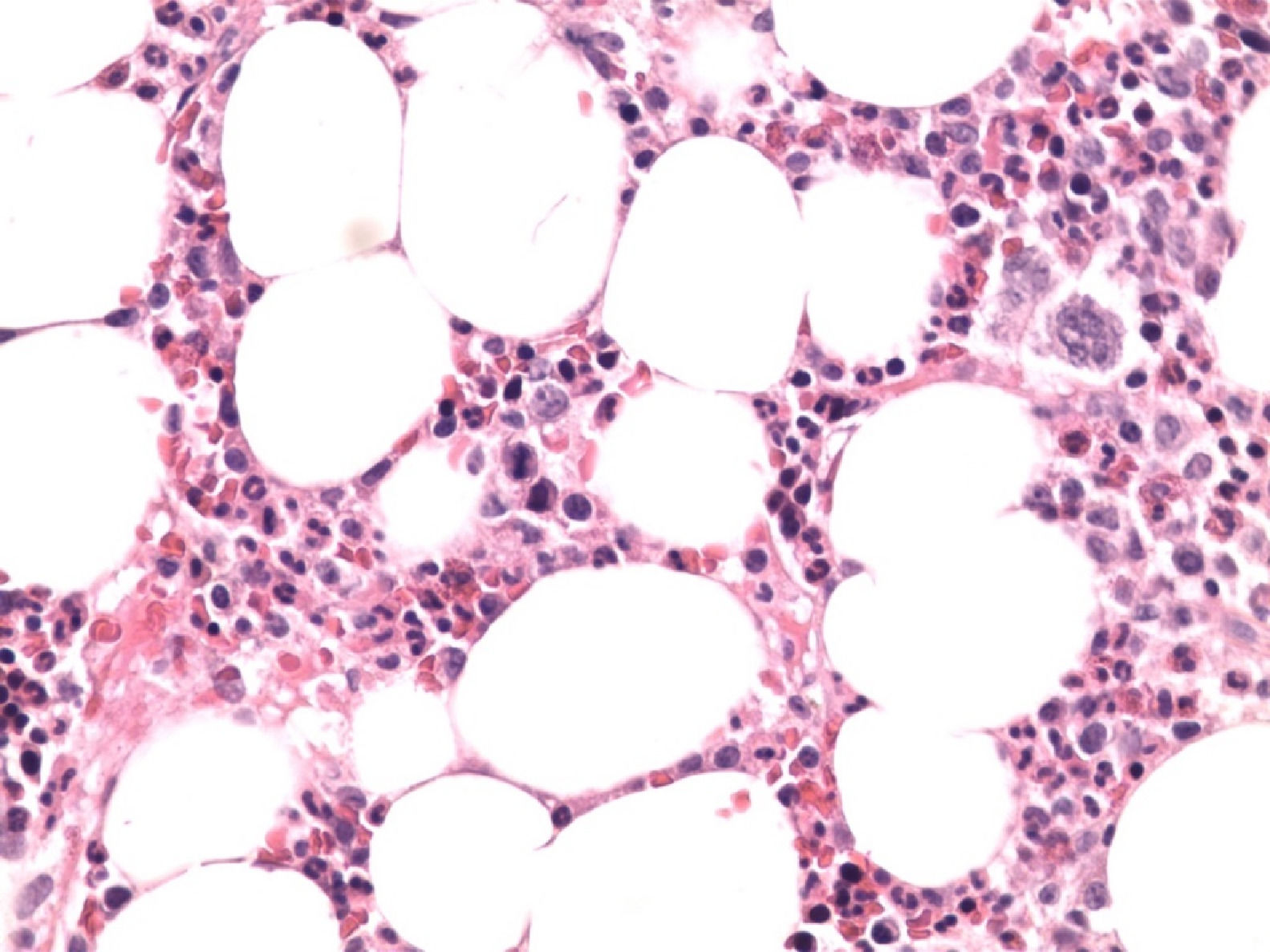
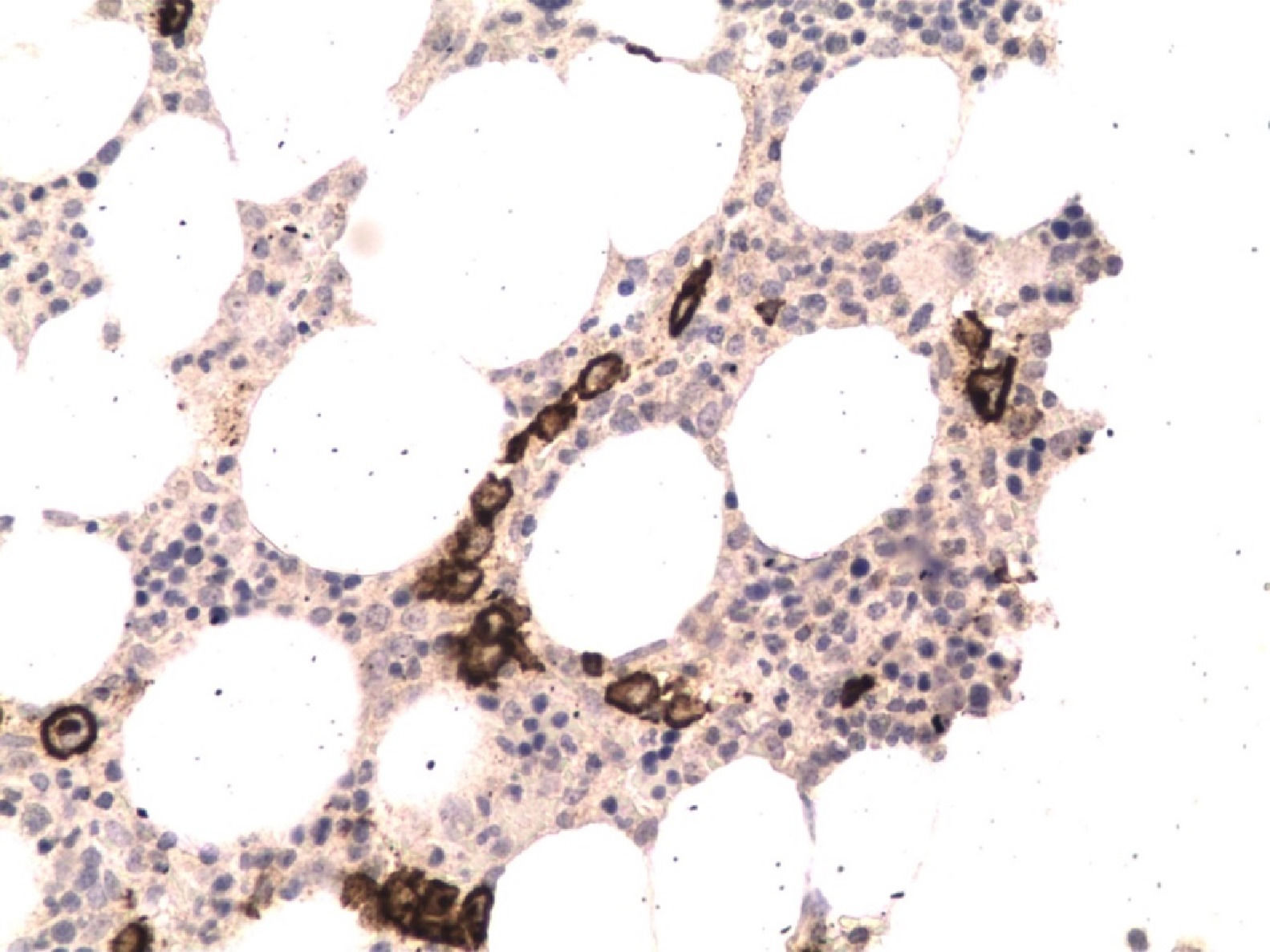
With these findings and due to the severity of the clinical situation, empirical treatment with HLH-94 protocol6 was started. Liver and renal dysfunction corrected, cytopenias improved, coagulation tests normalized, fever disappeared and ferritin levels decreased after 2 weeks of treatment and sIL-2r decreased to 1350 U/ml. After 3 weeks on treatment, fever reappeared, anemia (Hemoglobin 7,8 gr/dl) and ferritin of 3076 ng/mL were observed. At this point bone marrow biopsy was informed which showed infiltration by Intravascular Large B cell Lymphoma with no evidence of hemophagocytosis (Figures 1 and 2).


The patients started chemo-immunotherapy with R-CHOP regimen and intrathecal methotrexate as SNC prophylaxis. He received 6 cycles with normalization of laboratory parameters after 2nd cycle, achieving a complete remission at the end of treatment. He will receive high dose chemotherapy with autologous stem cell transplantation rescue as consolidation treatment.
DiscussionIntravascular large B cell lymphoma is an extremely rare subtype of extranodal large B-cell lymphoma characterized by the selective growth and infiltration by neoplastic lymphocytes in the lumina of small to medium sized blood vessels.
It has an aggressive behaviour associated with a poor outcome. Diagnosis of this entity poses a challenge to the hematologist and it is often delayed, which explains that historically more than half of the cases were diagnosed on autopsy. The data on this particular lymphoma, in terms of clinical presentation, treatment and outcomes come largely from case reports and small case series. The median age at diagnosis is 70 years (34–90) without sex prevalence. The most common symptom is fever of unknown origin, and a rapid deterioration of general condition is very frequent. Three main variants are described: classical, cutaneous and Hemophagocytic syndrome–associated.
Hemophagocytic lymphohistiocytosis (HLH) can develop secondary to hematologic malignancies, mainly T/NK lymphomas, being B-cell lymphomas a much rarer cause of this complication.7,8 The IVLBCL associated with hemophagocytic syndrome is the so called Asian variant and portends a dismal prognosis.5 The western variant typically presents with skin and CNS involvement and HLH is only exceptionally seen.9 There are very few reports of the Asian variant in patients from western countries in the literature with no cases reported in Latinoamerica.10 This entity has a median survival of 2–8 months in the pre Rituximab era. Treatment of choice is chemoimmunotherapy as in DLBCL. The addition of Rituximab has significantly changed the prognosis of IVLBCL. In western countries R-CHOP leads to CR in 88% and a 3-year OS rate of 81%. In Japan, 2-year progression-free survival and OS is 56% and 66% respectively. CNS is the most common site of relapse, therefore, prophylaxis is recommended. The role of Autologous Stem Cell Transplantation as consolidation is not well established, but it is an option in fit patients.
In summary, we present a case of a male patient who presented with fever of unknown origin, rapid development of multiorgan failure and very high levels of ferritin and soluble rIL-2. These findings led to the suspicion of HLH and the bone marrow biopsy confirmed the presence of IVLBCL. This variant has typically been described in Asian population, and as far as we know this is the first reported case in Latin America. This diagnosis represents a challenge, and in this particular case the IVLBCL was incidentally found as a result of a bone marrow biopsy performed in the diagnostic algorithm of fever of unknown origin highlighting the importance of this exam in the correct evaluation of patients with this clinica, presentation, and the need of a high level of suspicion of this entity in similar cases even in western countries.


