



Sickle cell anemia (SCA) is a Mendelian disorder with a heterogeneous clinical course. The reasons for this phenotypic diversity are not entirely established, but it is known that high fetal hemoglobin levels lead to a milder course of the disease. Additionally, genetic variants in the intergenic region HBS1L-MYB promote high levels of fetal hemoglobin into adulthood.
ObjectiveIn the present study, we investigated the HMIP1 C-839A (rs9376092) polymorphism, located at the HBS1L-MYB intergenic region block 1, in SCA patients.
MethodWe analyzed 299 SCA patients followed in two reference centers in Brazil. The HMIP1 C-839A (rs9376092) genotypes were determined by allele specific polymerase chain reactions. Clinical and laboratory data were obtained from patient interviews and medical records.
ResultsThe median fetal hemoglobin levels were higher in patients with the HMIP1 C-839A (rs9376092) AA genotype (CC=6.4%, CA=5.6% and AA=8.6%), but this difference did not reach significance (p=0.194). No association between HMIP1 C-839A (rs9376092) genotypes and other clinical and laboratorial features was detected (p>0.05).
ConclusionIn summary, our data could not support the previously related association between the HMIP1 C-893A (rs9376092) polymorphism and differential fetal hemoglobin levels.
Sickle cell anemia (SCA) is a monogenic disorder caused by the homozygosity of a missense mutation (Glu6Val, rs334) in the β-globin gene (HBB), resulting in HbS polymerization upon deoxygenation.1 Although SCA is a Mendelian disorder, the clinical course of the disease can be very heterogeneous. For instance, some subjects present most of the clinical and laboratory sub-phenotypes of the disease, wherefore they require hospitalization, whereas others show only mild symptoms and are followed during routine appointments.2 The reasons that account for the pathophysiological heterogeneity of SCA are not entirely elucidated and studies suggest that this variability may be dictated by genetic variants.3–6
The most well-established genetic modifier of SCA is the persistence of high fetal hemoglobin (HbF, α2γ2) levels into adulthood.7,8 While healthy individuals produce less than 1% of HbF, these values can range between 1% and 30% in SCA patients.1 High levels of HbF lead to a reduction in HbS concentration inside the red blood cells, preventing HbS polymerization and, subsequently, the formation of sickle cells.9 Variabilities in HbF concentrations can be traced back to the β-globin gene (HBB) cluster haplotypes,10 whereby the first genetic variant associated with increased HbF was the Senegal haplotype (rs7482144). In 10 to 50% of patients with SCA and persistence of high HbF levels into adulthood, genetic variants are also found in the BCL11A and MYB genes, which encode hematopoietic transcription factors.1,4 The latter gene is extensively associated with intergenic single nucleotide polymorphisms (SNPs) located on chromosome 6q23, also known as the HMIP2 (HBS1L-MYB intergenic polymorphism (HMIP) block 2). Recently, the downregulation of a long noncoding RNA (HMI-LNCRNA) transcribed from the enhancer region of MYB was associated with a significant increase in HbF levels.11 Additionally, a link between polymorphisms on HMIP2 and high levels of HbF was demonstrated, while their impact on stroke development, which can be a major complication in SCA, has also been reported.12 In contrast to HMIP2, the influence of the HMIP1 on HbF levels and SCA clinical course is still largely unknown. Hence, we evaluated the HMIP1 C-839A (rs9376092), which presented strong linkage disequilibrium with other variants in SCA patients and correlated these findings with clinical and laboratory data.
Material and methodsPatientsBetween December 2015 and February 2017, 299 adult patients with SCA and the βS-globin gene haplotype CAR/CAR were enrolled in this study. Patients were followed at two reference centers in Brazil (the Hematology and Hemotherapy Foundation of Pernambuco, Recife in northeastern Brazil and the State Institute of Hematology Arthur de Siqueira Cavalcanti, Rio de Janeiro in southeastern Brazil). The inclusion criteria stipulated that the patients had to be clinically stable for at least 3 months prior to blood collection, without signs of pain, vaso-occlusive crises or blood transfusions. Additionally, patients eligible for this study had not been receiving hydroxyurea therapy for at least 3 months before the inclusion, mainly due to personal reasons (procreation-related issues, side effects or drug-related intolerance) not related to the study, and remained without the use of hydroxyurea until the end of the study. Informed consent was obtained from all patients or their relatives, as appropriate, and clinical/laboratory data were obtained from patient interviews and medical records. The Institutional Research Ethics Board approved this study (#035/10) in accordance with the Declaration of Helsinki.
Genomic DNA extraction and allelic specific polymerase chain reactionGenomic DNA was extracted from peripheral blood leukocytes using the Puregene Kit (Gentra System, Minneapolis, MN, USA). The HMIP1 C-839A (rs9376092) was amplified by the allele-specific polymerase chain reaction (AS-PCR), with a total volume of 25μL under the following conditions: one cycle of denaturing, 96°C for 5min; 35 cycles of denaturing, 95°C for 30s; annealing, 58°C for 30s and extension, 72°C for 30s, and; a final cycle of extension, 72°C for 5min. The reaction was conducted using forward and reverse primers for internal control (F: 5ʹ-CTGCTACTGCCATCTGGGA-3ʹ; R: 5ʹ-CTGCACCAGTTACATGCCA-3ʹ) and specific primers for discriminating the alleles (F allele C: 5ʹ-GAAGACAGGCAGAATGAGAAAC-3ʹ; R allele A: 5ʹ-GGCCAACATTGTTCGTCTTTT-3ʹ). Therefore, the AS-PCR was performed in two independent reactions, one specific for each allele. The PCR products were subjected to gel electrophoresis in 1.5% agarose and the amplification products of internal control (305bp); allele C (241bp) and allele A (106bp) were visualized (Figure 1). To confirm these results, DNA sequencing was performed in at least 5% of random samples.
 for HMIP1 C-839A (rs9376092). The AS-PCR products were subjected to gel electrophoresis in 1.5% agarose. Lane 1 shows 1kb DNA-ladder (Invitrogen, Carlsbad, CA, USA). The pattern for the ancestral allele C (241bp) is present in lanes 2, 4, 6, 8 and 10. Lanes 3 and 13 exhibit the pattern for the variant allele A (106bp). The reaction internal control (305bp) is visualized in lanes 2–13. Patients 2, 3, 4 and 5 are homozygous for the CC genotype, while patients 1 and 6 present the CA and AA genotypes, respectively.")
Allele-specific polymerase chain reaction (AS-PCR) for HMIP1 C-839A (rs9376092). The AS-PCR products were subjected to gel electrophoresis in 1.5% agarose. Lane 1 shows 1kb DNA-ladder (Invitrogen, Carlsbad, CA, USA). The pattern for the ancestral allele C (241bp) is present in lanes 2, 4, 6, 8 and 10. Lanes 3 and 13 exhibit the pattern for the variant allele A (106bp). The reaction internal control (305bp) is visualized in lanes 2–13. Patients 2, 3, 4 and 5 are homozygous for the CC genotype, while patients 1 and 6 present the CA and AA genotypes, respectively.
Statistical analyses were performed using SPSS Statistics 19.0 (IBM Corporation, Somers, NY, USA) software. Fisher's exact test or Chi-square test, as appropriate, was used to compare categorical variables. Continuous variables were expressed as median and were compared by the Mann–Whitney or Kruskal–Wallis test, followed by Dunn's multiple comparisons post-test. All p-values were two-sided with a significance level of 0.05.
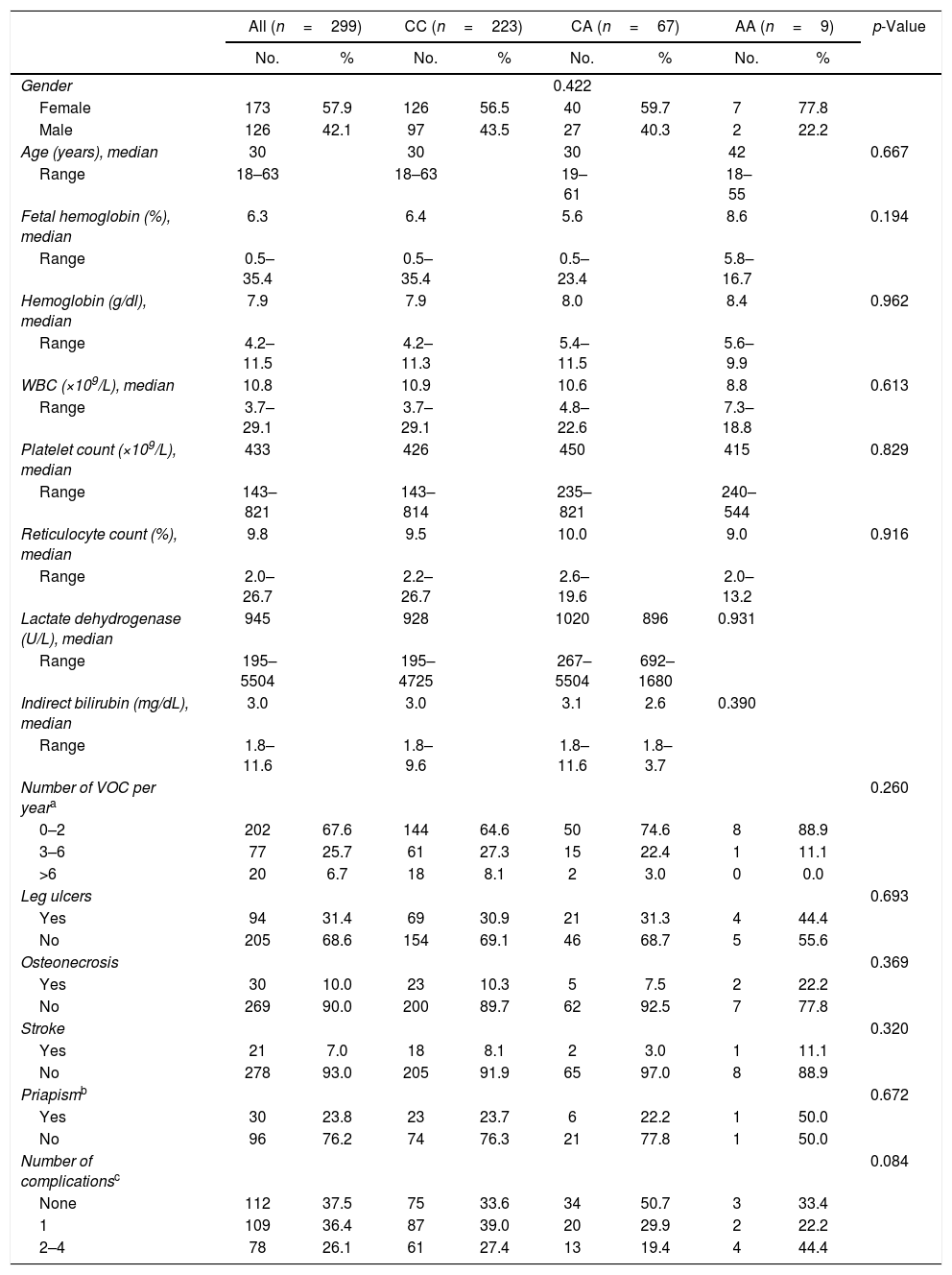
ResultsPatient characteristicsWith a median follow-up of 14 years (range: 1–34 years), the median age was 30 years (range: 18–63) and more than half of the individuals were females (173 out of 299, 57.9%). The main clinical and laboratory features are summarized in Table 1. Among the 299 patients, 94 (31.4%) developed leg ulcers, 30 (10.0%) osteonecrosis and 21 (7.0%) had strokes. Furthermore, over a fifth of the studied males had priapism (30 out of 126 males, 23.8%), while 112 (37.5%) patients had no clinical complications and 109 (36.4%) and 78 (26.1%) patients presented one or more clinical complications, respectively. Additionally, 202 patients had less than three vaso-occlusive crises (VOC) which required hospitalization per year in the last year (67.6%), while 77 (25.7%) and 20 (6.7%) patients had three to six and more than six VOC crisis per year, respectively.
Baseline characteristics of adult SCA patients according to HBS1L-MYB (rs9376092) genotypes.
| All (n=299) | CC (n=223) | CA (n=67) | AA (n=9) | p-Value | |||||
|---|---|---|---|---|---|---|---|---|---|
| No. | % | No. | % | No. | % | No. | % | ||
| Gender | 0.422 | ||||||||
| Female | 173 | 57.9 | 126 | 56.5 | 40 | 59.7 | 7 | 77.8 | |
| Male | 126 | 42.1 | 97 | 43.5 | 27 | 40.3 | 2 | 22.2 | |
| Age (years), median | 30 | 30 | 30 | 42 | 0.667 | ||||
| Range | 18–63 | 18–63 | 19–61 | 18–55 | |||||
| Fetal hemoglobin (%), median | 6.3 | 6.4 | 5.6 | 8.6 | 0.194 | ||||
| Range | 0.5–35.4 | 0.5–35.4 | 0.5–23.4 | 5.8–16.7 | |||||
| Hemoglobin (g/dl), median | 7.9 | 7.9 | 8.0 | 8.4 | 0.962 | ||||
| Range | 4.2–11.5 | 4.2–11.3 | 5.4–11.5 | 5.6–9.9 | |||||
| WBC (×109/L), median | 10.8 | 10.9 | 10.6 | 8.8 | 0.613 | ||||
| Range | 3.7–29.1 | 3.7–29.1 | 4.8–22.6 | 7.3–18.8 | |||||
| Platelet count (×109/L), median | 433 | 426 | 450 | 415 | 0.829 | ||||
| Range | 143–821 | 143–814 | 235–821 | 240–544 | |||||
| Reticulocyte count (%), median | 9.8 | 9.5 | 10.0 | 9.0 | 0.916 | ||||
| Range | 2.0–26.7 | 2.2–26.7 | 2.6–19.6 | 2.0–13.2 | |||||
| Lactate dehydrogenase (U/L), median | 945 | 928 | 1020 | 896 | 0.931 | ||||
| Range | 195–5504 | 195–4725 | 267–5504 | 692–1680 | |||||
| Indirect bilirubin (mg/dL), median | 3.0 | 3.0 | 3.1 | 2.6 | 0.390 | ||||
| Range | 1.8–11.6 | 1.8–9.6 | 1.8–11.6 | 1.8–3.7 | |||||
| Number of VOC per yeara | 0.260 | ||||||||
| 0–2 | 202 | 67.6 | 144 | 64.6 | 50 | 74.6 | 8 | 88.9 | |
| 3–6 | 77 | 25.7 | 61 | 27.3 | 15 | 22.4 | 1 | 11.1 | |
| >6 | 20 | 6.7 | 18 | 8.1 | 2 | 3.0 | 0 | 0.0 | |
| Leg ulcers | 0.693 | ||||||||
| Yes | 94 | 31.4 | 69 | 30.9 | 21 | 31.3 | 4 | 44.4 | |
| No | 205 | 68.6 | 154 | 69.1 | 46 | 68.7 | 5 | 55.6 | |
| Osteonecrosis | 0.369 | ||||||||
| Yes | 30 | 10.0 | 23 | 10.3 | 5 | 7.5 | 2 | 22.2 | |
| No | 269 | 90.0 | 200 | 89.7 | 62 | 92.5 | 7 | 77.8 | |
| Stroke | 0.320 | ||||||||
| Yes | 21 | 7.0 | 18 | 8.1 | 2 | 3.0 | 1 | 11.1 | |
| No | 278 | 93.0 | 205 | 91.9 | 65 | 97.0 | 8 | 88.9 | |
| Priapismb | 0.672 | ||||||||
| Yes | 30 | 23.8 | 23 | 23.7 | 6 | 22.2 | 1 | 50.0 | |
| No | 96 | 76.2 | 74 | 76.3 | 21 | 77.8 | 1 | 50.0 | |
| Number of complicationsc | 0.084 | ||||||||
| None | 112 | 37.5 | 75 | 33.6 | 34 | 50.7 | 3 | 33.4 | |
| 1 | 109 | 36.4 | 87 | 39.0 | 20 | 29.9 | 2 | 22.2 | |
| 2–4 | 78 | 26.1 | 61 | 27.4 | 13 | 19.4 | 4 | 44.4 | |
Abbreviations: WBC, White blood cells; VOC, vaso-occlusive crisis.
All patients were characterized for the HMIP1 C-893A (rs9376092) polymorphism. In our cohort, 223/299 patients (74.6%) carried the CC genotype, while 67/299 (22.4%) and 9/299 (3.0%) presented CA and AA genotypes, respectively.
The patients with SCA carrying the HMIP1 C-893A (rs9376092) AA genotype presented slightly increased, but not statistically significant (p=0.194, Figure 2), median HbF levels in comparison to the other genotypes (CC=6.4%, CA=5.6% and AA=8.6%). Although there were higher median levels of total hemoglobin and lower median levels of reticulocyte count, lactate dehydrogenase and indirect bilirubin in patients with the HMIP1 C-893A (rs9376092) AA genotype, no association between clinical/laboratory features and the HMIP1 C-893A (rs9376092) genotypes was detected (p>0.05).
Discussion levels of sickle cell anemia patients according to HMIP1 (rs9376092) genotypes. Median HbF levels were 6.9%, 5.7% and 8.4% for CC, CA and AA genotypes, respectively.")
Sustained postnatal expression of fetal hemoglobin is the main modulator of sickle cell anemia severity.9 The levels of HbF in SCA patients vary considerably, but appear to be constant after the age of 5 years.13,14 The HbF inhibits intracellular HbS polymerization and higher HbF levels are associated with reduced morbidity and mortality in patients with SCA.8 Hence, pharmacological induction of HbF is a logical approach to treat patients with SCA. Although hydroxycarbamide was shown to induce HbF in sickle cell disease over 30 years ago15 and its use is strongly encouraged due to its proven effectiveness,16,17 HbF induction can vary widely among patients on a similar daily dose.18–21 Considering the critical role of HbF in sickle cell anemia, it is pivotal to identify genetic modulators that impact HbF levels, in order to improve therapeutic strategies involving HbF induction.4,22,23
The intergenic region of HBS1L-MYB contains regulatory elements for MYB, which plays an important role in erythropoiesis.24 It is already well established that the overexpression/downregulation of MYB is associated with the inhibition/increment of HBG in adult erythroid cells.25,26 Therefore, the presence of polymorphisms in the HBS1L-MYB intergenic region, such as the rs9376092,27 affects the GATA transcription factor and thereby also MYB expression and HbF levels.27–30 Although polymorphisms in the HBS1L-MYB intergenic region have been associated with erythroid cell parameters, as red blood cells counts and mean corpuscular volume31,32 and HbF levels of patients with β-hemoglobinopathies,4,12,27,29,30 our results did not reveal any link between the HMIP1 C-839A (rs9376092) and clinical or laboratory features of SCA patients. To the best of our knowledge, our work is the first attempt to evaluate the impact of the HMIP1 C-893A (rs9376092) in a SCA context. However, we must bear in mind that only 3% of the patients enrolled in our study (9 out of 299) presented the HMIP1 C-839A AA genotype and that this low frequency may underestimate the real impact of this polymorphism on the HbF modulation.
The impact of HMIP2 polymorphisms on HbF levels appears to be more considerable in Brazilian patients, which is probably due to their genetic admixture,12 while further studies are still required to support this theory. In fact, this genetic admixture can also be the cause for the lack of association seen in our study. In Pernambuco and Rio de Janeiro, European admixture is marked in the general population33,34 and in SCA patients.35 Likewise, high admixture rates (65%) in sickle cell patients measured at the HBS1L-MYB locus was previously demonstrated.12 In this context, European chromosomes 6, in which the HMIP1 rs9376092-A is also more frequent, could have a large impact on our findings. Additionally, on European chromosomes, HMIP1 high-HbF variants are in a negative association with HMIP2 high-HbF variants.36 Thus, patients carrying the HMIP1 rs9376092-A are more likely to also carry the low-HbF genotype at HMIP2 as rs66650371 3bp insertion,37,38 negating the effect of the HMIP1 variant. Therefore, genetic admixture, in combination with the specific linkage disequilibrium at HBS1L-MYB, could be another reason for the lack of association seen in our study.
ConclusionHigh levels of fetal hemoglobin ameliorate the clinical outcome of patients with β-hemoglobinopathies and its induction has been a major focus of studies over the last decades. Although we demonstrated a lack of association between the HMIP1 C-893A (rs9376092) in two Brazilian cohorts, these results indicate that a set of environmental and genetic biomarkers can predict disease severity and that this prediction may vary for each population. Naturally, identifying these modulators is important for patients with β-hemoglobinopathies to develop new strategies for alternative therapies to improve the clinical outcome of SCA patients.
Author contributionsD.A.P-M. and I.F.D. performed experiments, analyzed and interpreted data and drafted the manuscript. E.B-J. and J.L.C-S. performed experiments, updated the clinical data and reviewed the manuscript. I.W. updated the clinical data and reviewed the manuscript. A.S.A., C.L.L., C.R.B-D. and M.A.B. recruited patients, updated the clinical data and reviewed the manuscript. A.R.L-A. analyzed and interpreted data, performed statistical analyses and reviewed the manuscript. D.A.P-M., J.L.C-S., and A.R.L-A. conceived and designed the study. A.R.L-A. gave the final approval of the version to be submitted.
Conflicts of interestThe authors declare no conflicts of interest.
The authors acknowledge all subjects and their parents for their cooperation in this study. This study was supported by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Grant # 483714/2013-5).


