


Studies have shown that the loss of the FOXO3 transcriptional function is involved in the pathophysiology of some chronic erythroid disorders, including beta-thalassemia (β-thal). Therefore, the single nucleotide polymorphism (SNP) rs3800231 (35-2764A > G) could contribute to alterations in its transcriptional activity, acting as a modifier of β-thal phenotypic manifestations.
Objective and methodIn order to better understand the genotypic and/or allelic distributions among β-thal patients, we evaluated 83 β-thal heterozygous and 20 homozygous, compared to 117 individuals without hemoglobinopathies (control group). Additionally, we verified any influence of the FOXO3 polymorphism on clinical manifestations among β-thal homozygotes.
ResultsWe obtained higher frequencies of the wild-type homozygous (AA) and the wild-type allele (A) in the β-thal group (p < 0.0001 and p = 0.00014, respectively). The most common clinical manifestations found among β-thal homozygotes were iron overload (90%), splenomegaly (65%) and bone complications (35%), e.g., osteopenia/osteoporosis. We observed that close to 80% of the patients presenting such manifestations had the genotype AA. However, we did not find any significant involvement of the FOXO3 polymorphism in clinical manifestation occurrences.
ConclusionThus, we concluded that the SNP rs3800231 did not play a significant role as a modifier of the clinical manifestations observed in the β-thal homozygotes studied.
Beta-thalassemia (β-thal) is an inherited monogenic disease, caused by mutations in the β-globin gene, characterized by the partial (β+) or total (β0) reduction of β-globin chains synthesis.1,2 The defect in the β-globin gene results in ineffective erythropoiesis, which leads to microcytic hypochromic anemia, a decrease in hemoglobin A synthesis, and excess of α-globin chains, the latter being responsible for the disease pathophysiology.3 As a consequence of these alterations, individuals may be clinically classified as β-thal major, resulting from homozygosis of β0 or β+ mutations, or in rare cases due to compound heterozygous (β0/β+), requiring chronic blood transfusions, as well as iron chelation therapy due to iron overload (IO).3 β-thal intermedia patients (β+/β+ or β0/β+), in which part of the homozygotes develops milder forms of the disease, ranging from moderate to severe anemia, requiring or not periodic transfusions.1,3 Finally, β-thal carriers, which are heterozygous (β0/β or β+/β), are usually asymptomatic.1 Thus, it is a complex group of diseases with a quite variable pathophysiology.
The FOXO3 gene encodes a protein that plays a key role in numerous cellular processes by controlling the transcription of several target genes. The FOXO3 activity investigation during erythropoiesis indicated fundamental functions for this protein in several steps, such as the maintenance of the hematopoietic stem cell (HSC) pool, cell cycle progression in the early-stage erythroblasts, erythroid maturation in the late-stage erythroblasts, correct temporal expression of genes responsible for the maturation and enucleation of erythroblasts, being also important in mitochondrial clearance due to autophagy (reviewed in reference4).
The FOXO3 knockout in mice has been described as causing a significant increase in reticulocytes, decreased red blood cell count, terminal erythroid maturation and defective erythrocyte production.5,6 There are also reports indicating that loss of the transcriptional function of the FOXO3 is involved in the pathophysiology of some chronic erythroid disorders, including β-thal.7 The loss of the transcriptional function may lead to an increase in myeloproliferation, immunological deficiencies and anemia due to failure in the transcriptional regulation of the antioxidant enzyme expressions, thus causing impairment in the control of the levels of reactive oxygen species in the HSCs (reviewed in reference4). Thus, the activity of FOXOs and the cellular redox state are intrinsically linked and the FOXO3 role can be accurately summarized as being a central component of resistance to cellular stress.8
Studies, including genome-wide association studies (GWAS), have been conducted relating the single nucleotide polymorphisms (SNPs), belonging to the FOXO3 gene, to human longevity and prevention of life-limiting diseases.9,10 The literature has shown that the polymorphism rs3800231 (35-2764A > G), located in the third intron of this gene, is related to gender-independent longevity9,10 and has never been studied in hematological diseases. As demonstrated, the mechanisms that guarantee the correct transcriptional regulation of the FOXO3 gene are essential for its activity to trigger appropriate cellular responses. Thus, the presence of the aforementioned FOXO3 polymorphism could contribute to alterations in the transcriptional activity of this gene, acting as a modifier of β-thal phenotypic manifestations, according to its great relevance in the erythropoiesis process.
Materials and methodsSubjectsThe cohort was composed of 220 unrelated individuals from southeastern Brazil, divided into three groups. The control group consisted of 117 individuals without hemoglobinopathies (69 women and 48 men, mean age: 29.4 ± 12.12). The β-thal group was composed by 83 heterozygotes (54 women and 29 men; mean age: 32.79 ± 20.32), and 20 homozygotes (8 women and 12 men, mean age: 29 ± 15) in a clinical follow-up at the hospitals located in São Carlos, Sao Paulo and the State Institute of Hematology Arthur de Siqueira Cavalcanti (HEMORIO), Rio de Janeiro, Brazil, respectively. Among the homozygotes, most individuals (90%) were on a regular blood transfusion regimen (intervals of 15 or 21 days) and 18 (90%) of them were undergoing iron chelation therapy. None of the patients was using hydroxyurea (HU). All patients had access to the same medication protocol.
Moreover, all subjects gave their informed consent and the Data Safety Monitoring Board (DSMB), in compliance with Brazilian regulations, approved the study. In addition, we obtained clinical data from the β-thal homozygotes by reviewing each patient's medical record from the Blood Center database, under the supervision of clinicians responsible for the patients.
Biological samplesBlood samples (about 8 mL) were collected through venipuncture in EDTA tubes. We used four milliliters (mL) of whole blood for cytological, electrophoretic and chromatographic hemoglobin (Hb) identification tests. The other fraction of whole blood was used for DNA extraction from leukocytes for further molecular analysis.
Hemoglobin screening testsThe cell morphology microscopic analysis was performed on the stained blood, using the May-Grünwald-Giemsa under light microscopy with a 40× objective. The Hb phenotype identification was performed using electrophoresis on cellulose acetate with a pH of 8.6, and agar electrophoresis with a pH of 6.2 (reviewed in reference11). The Hb fraction quantification was obtained using high-performance liquid chromatography (HPLC) in the automated ultra2 Resolution™ (Trinity Biotech) system.
Molecular analysisThe DNA was extracted according to Sambrook et al.12 The β-thal genotyping was performed by AS-PCR, investigating the most frequent β-thal mutations in Brazil (IVS-I-1, IVS-I-6, IVS-I-110, and codon 39).13 When the mutational profile was not defined, DNA sequencing was performed. We investigated mutations in several regions of the β-globin gene (promoter region, exons, introns and untranslated regions), equaling an 1825 bp fragment. The PCR products were evaluated by direct genomic sequencing by the automated sequencer ABI PRISMTM 3500xl Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). Sequences were evaluated using the BioEdit Sequence Alignment Editor package (Ibis Biosciences, Carlsbad, CA, USA), Phred/Phrap/Consed (University of Washington, Seattle, WA, USA; http://ww.phrap.org/) and the alignment of the sequences was performed using the ClustalW multiple sequence alignment program,14 associated with the Bioedit package (http://www.mbio.ncsu.edu/bioedit/bioedit.html). The clinical classification was determined according to the type of mutation detected and the phenotype presented.
The FOXO3 polymorphism (35-2764A > G) investigation was assessed using the PCR-RFLP with specific primers and the amplicon was cleaved in all subjects by the RsaI (New England BioLabs, MA, USA) restriction endonuclease.10
Statistical analysisTo compare the genotypes of the SNP rs3800231 of the FOXO3 gene between the groups studied, we performed Pearson's χ2 test, complemented by Fisher's exact test in Statistica.9 0 software (Statsoft Inc. Tulsa, OK, USA). Finally, to evaluate the odds ratio (retrospective group) for the occurrence of the most frequent clinical manifestations (dependent variable) among the homozygous beta-thalassemia group, according to age, gender and genetic polymorphisms evaluated (independent variables), we performed a multiple logistic regression (MLR) using the software BioEstat.Ink 5.0, which presents the result for the odds ratio from a binominal analysis (0 = failure, 1 = success). The level of significance was set at p < 0.05.
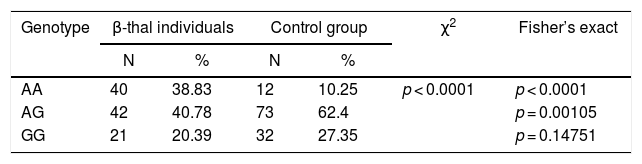
ResultsThe genotypic and allelic frequency for polymorphism rs3800231Among the 117 control subjects evaluated, we identified 12 (10.25%) wild homozygotes (AA), 73 (62.4%) heterozygotes (AG) and 32 (27.35%) homozygous mutants (GG) for the SNP rs3800231, leading to an allelic frequency of 0.41 for the wild-type allele (A) and 0.59 for the (G) mutant. Regarding the thalassemic group, for the 83 β-thal heterozygotes, we identified 24 AA (28.91%), 40 AG (48.19%) and 19 GG (22.9%), resulting in an allele frequency of 0.53 for allele A and 0.47 for G. Finally, for the 20 β-thal homozygotes, we identified 16 AA (80%), 2 AG (10%) and 2 GG (10%), leading to an allelic frequency of 0.85 for allele A and 0.15 for G. Due to the low sample size of the FOXO3 mutants in the β-thal homozygotes, we compared the FOXO3 genotypes between control and thalassemic groups (Table 1).
Comparison of genotypic and allelic frequencies of rs3800231 polymorphism between thalassemic and control groups.
| Genotype | β-thal individuals | Control group | χ2 | Fisher’s exact | ||
|---|---|---|---|---|---|---|
| N | % | N | % | |||
| AA | 40 | 38.83 | 12 | 10.25 | p < 0.0001 | p < 0.0001 |
| AG | 42 | 40.78 | 73 | 62.4 | p = 0.00105 | |
| GG | 21 | 20.39 | 32 | 27.35 | p = 0.14751 | |
| Allele | β-thal individuals | Control group | χ2 | Fisher’s exact | ||
|---|---|---|---|---|---|---|
| N | % | N | % | |||
| A | 122 | 59.2 | 97 | 41.5 | p = 0.00020 | p = 0.00014 |
| G | 84 | 40.8 | 137 | 58.5 | ||
Comparisons were made by the Pearson Chi-square test, supplemented by Fisher's exact.
test. N = sample number; %: frequency. In bold: significant statistical value.
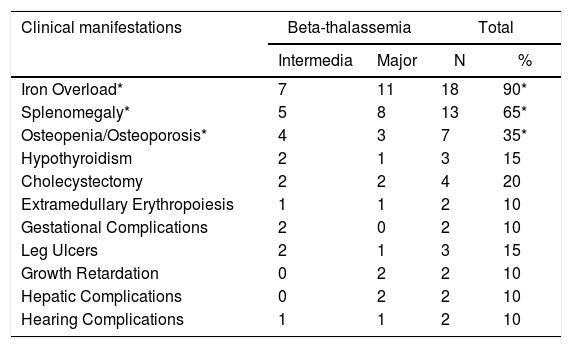
Among the different clinical manifestations reported by the 20 homozygous β-thal patients, the most common was iron overload (IO) [18 (90%) individuals], followed by splenomegaly [13 (65%)] and bone complications, e.g., osteopenia and osteoporosis (OO) [7 (35%)], as can be observed in Table 2.
Clinical manifestations observed among homozygous β-thalassemic patients.
| Clinical manifestations | Beta-thalassemia | Total | ||
|---|---|---|---|---|
| Intermedia | Major | N | % | |
| Iron Overload* | 7 | 11 | 18 | 90* |
| Splenomegaly* | 5 | 8 | 13 | 65* |
| Osteopenia/Osteoporosis* | 4 | 3 | 7 | 35* |
| Hypothyroidism | 2 | 1 | 3 | 15 |
| Cholecystectomy | 2 | 2 | 4 | 20 |
| Extramedullary Erythropoiesis | 1 | 1 | 2 | 10 |
| Gestational Complications | 2 | 0 | 2 | 10 |
| Leg Ulcers | 2 | 1 | 3 | 15 |
| Growth Retardation | 0 | 2 | 2 | 10 |
| Hepatic Complications | 0 | 2 | 2 | 10 |
| Hearing Complications | 1 | 1 | 2 | 10 |
The genotypic frequencies observed for the SNP among the patients presenting the most frequent clinical manifestations were 14 (77.78%) AA, two (11.11%) AG and two (11.11%) GG for IO; 11 (84.6%) AA, one (7.7%) AG and one (7.7%) GG for splenomegaly, and; six (85.7%) AA and one (14.3%) GG for OO. In other words, we showed that wild homozygotes (AA) were the most common FOXO3 genotype (∼80%) found among these patients, raising the hypothesis that this genotype could influence the occurrence of clinical subphenotypes in β-thalassemic patients.
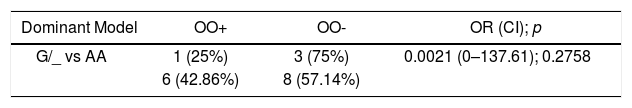
Moreover, extensive iron deposition leads to bone marrow cavity expansion and reduced trabecular bone volume, resulting in decreased bone tissue and osteopenia/osteoporosis.15 Taking into consideration this close relationship between the IO and bone complications, along with the fact that all individuals with bone complications also presented an IO in our study, we performed a Multiple Logistic Regression (MLR). We tested if the FOXO3 polymorphism investigated was related to the chance of presenting bone complication within the group that is already known to have IO, in both genetic recessive and dominant models. Unexpectedly, we did not find any relationship between the polymorphism presence and bone complications in β-thal homozygotes with IO (Table 3). However, bone complications present a multifactorial character in β-thal homozygotes, such as the severity of anemia, adequacy of transfusion therapy and chelation, level of the IO, endocrine alterations (e.g., hypogonadal thalassemia patients have lower bone mineral density (BMD)), micronutrient deficiency (e.g., zinc) and renal impairment (e.g., there is an association between hypercalciuria and lower BMD in β-thal patients, as reviewed in reference16). Thus, the occurrence of bone complications could be explained by changes in some of the other factors mentioned above.
Relative risk assessment for the frequency of the SNP genotype among individuals with iron overload, with or without bone complications.
| Dominant Model | OO+ | OO- | OR (CI); p |
|---|---|---|---|
| G/_ vs AA | 1 (25%) | 3 (75%) | 0.0021 (0–137.61); 0.2758 |
| 6 (42.86%) | 8 (57.14%) |
| Recessive Model | OO+ | OO- | OR (CI); p |
|---|---|---|---|
| GG vs A/_ | 1 (50%) | 1 (50%) | 4.2078 (0.03–606.87); 0.2758 |
| 6 (37.5%) | 10 (62.5%) |
Test performed: Multiple Logistic Regression, considering having bone complication = 1 vs. not having = 0. Dominant model: the mutant homozygote and heterozygous homozygotes (GG + AG = success) are evaluated for the SNP versus wild homozygote (AA = failure). Recessive model: the mutant homozygote (GG = success) is evaluated for the SNP versus wild and heterozygous homozygotes (AA + AG = failure). OO +: individuals with osteopenia/osteoporosis. OO-: individuals without osteopenia/osteoporosis. OR: odds ratio. CI: confidence interval.
It is also known that splenomegaly, a common result of ineffective erythropoiesis in β-thal patients, occurs in FOXO3-deficient mice, due to an increase in extramedullary hematopoiesis containing erythroid and myeloid cells in the spleen and liver.17 Therefore, we investigated the possible influence of the FOXO3 polymorphism on the chance of having splenomegaly, in both the recessive and dominant models. As with the bone complication results, we did not corroborate our hypothesis (Table 4).
Multiple Logistic Regression (MLR) assessment for the chance of having splenomegaly, related to the presence of the SNP in homozygous thalassemia.
| Dominant Model | + | – | OR (CI); p |
|---|---|---|---|
| G/_ vs. AA | 2 (50%) | 2 (50%) | 0.4921 (0.04–5.65); 0.5691 |
| 11 (68.75%) | 5 (31.25%) |
| Recessive Model | + | – | OR (CI); p |
|---|---|---|---|
| GG vs. A/_ | 1 (50%) | 1 (50%) | 0.5507 (0.03–10.63); 0.6928 |
| 12 (66.7%) | 6 (33.3%) |
Test performed: Multiple Logistic Regression, considering having splenomegaly = 1 vs. not having = 0. +: individuals with splenomegaly. -: individuals without splenomegaly. OR: odds ratio. CI: confidence interval.
The molecular defects that result in thalassemia are extremely diverse, with more than 350 existent β-thal mutations reported in the IthaGenes database.2 Therefore, the genetic and physiopathological understanding of the basis for disease expression variability is still unclear. The FOXO3 transcription factor is the main active FOXO within HSCs, acting as a key mediator of erythroid terminal maturation, enucleation and regulation of the antioxidant transcriptional response program.4 Recently, it has been demonstrated that the loss of FOXO3 activity is one of the causes of dysregulated erythropoiesis in β-thal cells.7
In a study with β-thal intermedia mice, the process of ineffective erythropoiesis was demonstrated to be occasioned by the inactivation of the FOXO3 that led to oxidative damage in late erythroblasts. This downregulation of the FOXO3 is due to persistent activation of the EPOR-PI3K/AKT/mTOR pathway,18 suggesting that the activation of the FOXO3 could be beneficial in this blood disorder and thus, different FOXO3 activating agents are being tested in vitro and animal models, such as resveratrol and rapamycin. Resveratrol administration increased Hb levels, reduced the reticulocyte count and improved erythrocyte survival,19 while rapamycin enhanced γ-globin mRNA expression, HbF production,20 erythroid cell maturation and β-globin production,18 improving the anemia outcome through FOXO3 activation. Thereby, genetic polymorphism effects on the FOXO3 gene expression have the potential to improve our understanding of β-thal pathophysiology and ineffective erythropoiesis.
The FOXO3 polymorphism rs3800231 reveals an extensive ethnic and geographic variability, showing an interesting association with human longevity but with contradictory results.10,21,22 This SNP presents a global minor allele frequency (MAF) of A = 0.46 and G = 0.53, while European populations present A = 0.32 and G = 0.67. In this study, we observed that the allele frequencies in the control group (A = 0.41 and G = 0.59) approach the global MAF, which is to be expected, due to the great miscegenation that occurred in the country. However, unexpectedly, we observed that the allelic frequencies observed among the β-thalassemic individuals (A = 0.59 and G = 0.41) differ greatly from those observed in the European population. This finding stands out because southeastern Brazil received a large number of immigrants of Mediterranean origin.23 Despite its widespread worldwide distribution, thalassemia has been known for many years as "Mediterranean Anemia" because of its high prevalence in individuals whose ancestors originated from Italy, Greece, and Armenia.2 These novel findings regarding the SNP allele frequencies, never before assessed in a hematological disorder, differ from those expected, given the ethnic relationship between β-thal and Europe. Thus, this polymorphic variant is an intriguing target in the study of this hematological disease, considering the essential function of the FOXO3 gene in erythropoiesis.
The characteristic ineffective erythropoiesis found in β-thalassemic patients triggers a cascade of compensatory mechanisms, resulting in different clinical manifestations.1 In this study, we have shown that the most frequent manifestations in our homozygous study group were IO, splenomegaly, and bone complications, e.g., osteopenia and osteoporosis. Interestingly, different findings in the scientific literature have related, directly or indirectly, these three clinical manifestations with the FOXO3 differential expression, as well as with the SNP rs3800231. In addition, the present study has observed a close relationship between the IO and bone complications, since all individuals with bone complications also presented IO. The IO damages the balance of the osteoblast metabolism and induces apoptosis in vitro, which may be the main reason for bone loss and osteoporosis.24 In addition, researchers demonstrated that the IO significantly threatened the growth of osteoblastic cells and that the FOXO3 binds directly to the DUSP14 gene promoter, whose overexpression significantly suppressed the effect of the IO in the osteoblastic cells, thus indicating that the interaction of the FOXO3 and DUSP14 may be responsible for the cellular defense under stress by the IO in these cells.25
Moreover, regarding bone complications, studies have shown that the FOXO3 is the predominant isoform expressed in bone cells.26 Loss of the transcriptional activity of the FOXO3 by gene silencing leads to an increased apoptosis of osteoblastic cells and a decreased number of osteoblasts and bone formation rate and bone mass.27,28 It is also known that the loss of FOXOs (FOXO1, 3, and 4) function in mice resulted in increased apoptosis of osteoblasts and osteocytes, resulting in an osteoporotic phenotype, characterized by decreased bone mass in the cortical and spongy regions. Thus, the function of the FOXO was considered indispensable to bone mass homeostasis.26 Additionally, FOXO3 levels are elevated during osteoblast differentiation, corresponding to an increased capacity of the human mesenchymal stem cells (hMSCs) to deal with oxidative stress, which is essential for their proper differentiation to osteoblasts.29 In addition, comparisons made between aging-related characteristics with the presence of 15 SNPs of the FOXO3 in the elderly of Danish origin found a significant reduction in the risk of bone fracture for the recessive alleles of 10 of these SNPs, among them the G allele of the SNP rs3800231.30
The FOXO3 protein levels are abundantly high in the spleen.9 In a mice study, those with a FOXO3 deficiency exhibited splenomegaly, increased splenocyte numbers, and extramedullary hematopoiesis, with a higher frequency of erythrocytic and granulocytic lines. In addition, the bone marrow was hypocellular, with a decreased production of mature erythroid cells, these findings being further corroborated by histopathological analysis.17 Therefore, these observations, along with the significant differences in genotypic and allelic frequencies found between control and thalassemic groups, the high frequency of wild homozygotes (AA) among the β-thal homozygotes presenting IO, splenomegaly and bone complications, were very suggestive that the FOXO3 polymorphism could influence the chance of clinical condition development.
ConclusionWe concluded that the FOXO3 polymorphism investigated did not play a significant role as a modifier of the clinical manifestations observed in the β-thal homozygotes studied. However, it is worth mentioning that the small sample size in some subgroups might have influenced our findings. Thus, taking into consideration the studies summarized above, the hypothesis postulated in this study is still plausible, suggesting that the SNP rs3800231, as well as others in the FOXO3 gene, might be involved in the broad spectrum of clinical subphenotypes of the individuals with beta-thalassemia.
AuthorshipF.F.T. and V.S.B.: study concept and design; data analyses and interpretation and manuscript preparation.
D.G.H.S.: data analyses and interpretation and critical review of the manuscript.
J.V.O.: data statistical analysis.
C.R.B.D.: study concept and design.
Conflicts of interestThe authors declare no conflicts of interest.
We would like to thank Clarisse Lopes de Castro Lobo (HEMORIO), Isabeth da Fonseca Estevão (Department of Medicine, Federal University of São Carlos – UFSCAR) and Angelica Marta Lopes (Department of Biochemistry and Molecular Biology, São José do Rio Preto Medical School – FAMERP) for providing samples. This research did not receive any specific grant from funding agencies in the public, commercial or not-for-profit sectors.


