

Myelodysplastic syndromes (MDS) comprise a group of malignant clonal hematologic disorders characterized by ineffective hematopoiesis and propensity for progression to acute myeloid leukemia. Acquired mutations in the gene encoding RNA splicing factor 3B subunit 1 (SF3B1) are highly associated with the MDS subtypes presenting ring sideroblasts, and represent a specific nosological entity. The effects of these mutations on clinical outcomes are diverse and contrasting.
MethodsA cohort of 91 Brazilian MDS patients, including patients with ring sideroblasts in the bone marrow, were screened for mutations in the SF3B1 hotspots (exons 12–15) by direct Sanger sequencing.
ResultsSF3B1 heterozygous mutations were identified in six patients (7%), all of them with ring sideroblasts, thus confirming the association between SF3B1 mutations and myelodysplastic syndrome subtypes bearing this morphologic feature (frequency of 6/13, p-value<0.0001).
ConclusionThis is the first screening of SF3B1 mutations in a cohort of Brazilian myelodysplastic syndrome patients. Our findings confirm that mutations in this splicing gene correlate with bone marrow ringed sideroblasts.
Myelodysplastic syndromes (MDS) are defined as clonal hematopoietic stem cell disorders characterized by cytopenia, myelodysplasia, ineffective hematopoiesis, and propensity for progression to acute myeloid leukemia.1–4 The MDS diagnosis is arrived at after exclusion of other myeloid neoplasms such as acute myeloid leukemia (AML), chronic myelomonocytic leukemia (CML), and myelodysplastic/myeloproliferative neoplasms. Morphologic criteria for diagnosis include dysplasia present in at least 10% of cells of any myeloid lineage with one or more affected lineages of the bone marrow.5 The dysplastic changes observed in the myeloid lineages probably arise from a genetically transformed, primitive hematopoietic stem cell, followed by other genetic and epigenetic changes, as well as stromal and immune responses in the host, that contribute to phenotypic diversity, hematopoietic efficiency, and susceptibility to leukemic transformation.6 MDS is categorized in six subtypes, according to the World Health Organization (WHO) criteria revised in 20087: refractory cytopenia with unilineage dysplasia (including refractory anemia, neutropenia, and thrombocytopenia), refractory cytopenia with multilineage dysplasia, refractory anemia with excess of blasts, MDS with isolated del(5q), unclassifiable MDS, and refractory anemia with ring sideroblasts (RS). In the latter, RS (perinuclear ring of granules formed by iron-loaded mitochondria) are observed in ≥15% of bone marrow erythroid precursors. Large numbers of RS are observed in the bone marrow of more than a quarter of patients with MDS,8 highlighting the importance of this subgroup in the disease.
The incidence of MDS increases at age 70 years and older9 and the direct correlation with age suggests genetic damage caused by hazardous exposure, e.g. chemotherapy, ionizing radiation, diesel fuel, and smoking.2,5 A wide diversity of mutations in individual genes has been described, suggesting that several molecular mechanisms are involved in disease development.10 Whole-exome sequencing studies identified recurrent somatic mutations in spliceosome complex genes in a significant proportion of adult MDS patients, with emphasis on the SF3B1 (splicing factor 3b, subunit 1) gene, mutated in about 20% of patients.3,11–13 High frequencies were especially observed in patients whose disease is characterized by the presence of RS, in which SF3B1 mutations were detected in 65%,11 75%,3 and 56%10 of patients.
The SF3B1 gene encodes the subunit 1 of the splicing factor 3b protein complex, which forms the U2 small nuclear ribonucleoprotein complex (U2 snRNP) along with splicing factor 3a and a 12S RNA unit. This complex plays a critical role in the splicing machinery and regulates the diversity of splice variants.14SF3B1 is also involved in the alternative splicing program of genes that control cell cycle progression and apoptosis, which may elucidate the critical contribution of mutant SF3B1 in modulating proliferation and survival of tumor cells.15 The SF3B1 protein is ubiquitously and highly expressed in hematopoietic lineages.16 The carboxy-terminal two-thirds of the subunit 1 has 22 non-identical, tandem HEAT domain (HD) repeats, and the majority of mutations in MDS are clustered in the region encoding the imperfect HD 4–9,16 encompassing exons 12–15 (codons 513–741). Mutations are usually somatic de novo, and often corresponding to missense nucleotide changes clustered mainly in three hotspots: codons 662, 666, and 700.15
In this study, 91 Brazilian MDS patients were screened for mutations in the SF3B1 hotspots to observe the heterogeneity, and the incidence in a Brazilian population.
MethodsSamples, DNA extraction, and polymerase chain reactionsPatients seen at two tertiary hematology centers in university hospitals located in two separate regions in the country were evaluated: one in the southeast (Ribeirão Preto, São Paulo) and the other in northeast of Brazil (Fortaleza, Ceará) that are more than 2600km apart. The populations in the regions where the two centers are located are socially, economically, and ethnically diverse. Blood samples were collected from 91 patients with MDS diagnosed and classified according to the WHO criteria (2008).7 Forty-five MDS patients seen between 2009 and 2011 at the hematology clinic, Hospital das Clínicas de Ribeirão Preto, Universidade de São Paulo, and from whom DNA samples were previously collected and available for testing were included in the study after MDS diagnosis was confirmed. Forty-six consecutive MDS patients seen at the hematology clinic, Universidade Federal do Ceará, diagnosed between 2008 and 2010 were also enrolled. Samples were collected and stored after patients had given their informed written consent and the study had been approved by the Local Ethics Committee (CEP-HCRP n. 7147/2005). The study was conducted in accordance with the Helsinki Declaration, as revised in 2008.
Total genomic DNA was extracted from blood leukocytes using TRIzol reagent (Ambion, Foster City, USA). All DNA samples were amplified by polymerase chain reaction (PCR) using specific primers for exons 12–15 of the SF3B1 gene (RefSeq NM_012433.2) and Platinum Taq DNA Polymerase kit (Invitrogen, Carlsbad, USA) according to the manufacturer's instructions. Primer sequences used in amplification reactions were designed according to Malcovati et al.17 The quality of PCR products was assessed by denaturing 1.5% agarose gel electrophoresis under standard conditions. The PCR products were purified using the QIAquick PCR Purification kit (Qiagen, Hilden, Germany) according to the manufacturer's recommendations.
Sanger sequencing and analysisPurified PCR fragments were directly sequenced in both directions. Sequencing reactions were performed using the DYEnamic ET Dye Terminator Cycle Sequencing kit and the MegaBACE Long Read Matrix equipment (GE Healthcare Life Sciences, Uppsala, Sweden). Chromatograms were analyzed through the CLC Main Workbench software v5.7 (CLCbio, Katrinebjerg, Denmark). The PolyPhen-2.0 tool18 was used to predict the impact of the detected amino acid substitutions on the structure and function of SF3B1 human protein.
Statistical analysisStatistical analysis was performed in the R software environment (http://www.r-project.org/). Pairwise comparisons were performed by Mann–Whitney test, and by two-sided Fisher exact test (CI=0.95) for quantitative and categorical variables, respectively.
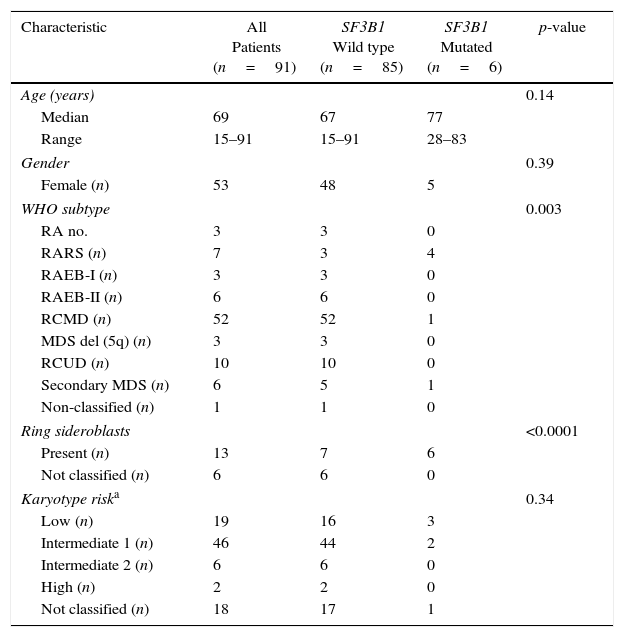
ResultsSanger sequencing was performed in 91 Brazilian MDS patients and three non-synonymous SF3B1 heterozygous mutations were identified, leading to missense substitutions in six unrelated patients. The most common recurrent SF3B1 mutation affected codon 700 (K700E, four out of six patients); one patient harbored a mutation in codon 622 (E622D), and another one, in codon 666 (K666R) (Figure 1A). The prediction of mutation malignancy or mildness showed mutations as probably damaging for the protein structure and function, with high scores, ranging from 0.87 to 0.99 (Figure 1B). Considering WHO subtypes, differences were detected comparing wild type and mutated groups (p-value=0.003; Table 1). A strong association between SF3B1 mutation and disease phenotype with RS (p-value<0.0001) was observed in this cohort. Thirteen patients (14.2%) were found with RS and all six SF3B1 mutated patients (6/13) presented at least 15% of RS in the bone marrow. The correlation of SF3B1 mutations in MDS with RS, the median age, gender, karyotype risk comparisons, and clinical characteristics of patients are shown in Table 1.
 Chromatograms of regions of exons 14 and 15 presenting nucleotide substitutions that lead to amino acid changes in the SF3B1 protein; one patient exhibited an E622D substitution, another one, a K666R, and four patients presented the most common described substitution, K700E. (B) Representation of the impact of each amino acid substitution on the protein structure and function, as well as a predicted score ranging from 0 to 1, in which 1 represents the higher probability of damaged protein.")
Heterozygous mutations in SF3B1 found in six MDS patients. (A) Chromatograms of regions of exons 14 and 15 presenting nucleotide substitutions that lead to amino acid changes in the SF3B1 protein; one patient exhibited an E622D substitution, another one, a K666R, and four patients presented the most common described substitution, K700E. (B) Representation of the impact of each amino acid substitution on the protein structure and function, as well as a predicted score ranging from 0 to 1, in which 1 represents the higher probability of damaged protein.
Clinical characteristics of 91 MDS patients according to SF3B1 mutation status.
| Characteristic | All Patients (n=91) | SF3B1 Wild type (n=85) | SF3B1 Mutated (n=6) | p-value |
|---|---|---|---|---|
| Age (years) | 0.14 | |||
| Median | 69 | 67 | 77 | |
| Range | 15–91 | 15–91 | 28–83 | |
| Gender | 0.39 | |||
| Female (n) | 53 | 48 | 5 | |
| WHO subtype | 0.003 | |||
| RA no. | 3 | 3 | 0 | |
| RARS (n) | 7 | 3 | 4 | |
| RAEB-I (n) | 3 | 3 | 0 | |
| RAEB-II (n) | 6 | 6 | 0 | |
| RCMD (n) | 52 | 52 | 1 | |
| MDS del (5q) (n) | 3 | 3 | 0 | |
| RCUD (n) | 10 | 10 | 0 | |
| Secondary MDS (n) | 6 | 5 | 1 | |
| Non-classified (n) | 1 | 1 | 0 | |
| Ring sideroblasts | <0.0001 | |||
| Present (n) | 13 | 7 | 6 | |
| Not classified (n) | 6 | 6 | 0 | |
| Karyotype riska | 0.34 | |||
| Low (n) | 19 | 16 | 3 | |
| Intermediate 1 (n) | 46 | 44 | 2 | |
| Intermediate 2 (n) | 6 | 6 | 0 | |
| High (n) | 2 | 2 | 0 | |
| Not classified (n) | 18 | 17 | 1 | |
RA: refractory anemia; RARS: refractory anemia with ring sideroblasts; RAEB-I, II: refractory anemia with excess blasts; RCMD: refractory cytopenia with multilineage dysplasia; MDS del (5q): MDS with isolated 5q deletion; RCUD: refractory cytopenia with unilineage dysplasia.
This study found that in a Brazilian cohort of MDS patients, SF3B1 is mutated in about 7% of patients, all of whom have RS in the bone marrow. The current diagnostic criteria (WHO, 2008) for MDS are mainly based on morphology4 but Malcovati et al.19 demonstrated the feasibility of including a molecular classification in myeloid neoplasms. The inclusion of molecular analysis might be valuable not just for diagnosis, but also for prognostic purposes and evaluation of disease progression. Therefore, the importance of the mutational analysis improves clinical decision making, as different point mutations in MDS have distinct effects on disease phenotype and prognosis.19,20 Apart from the WHO category, SF3B1 mutation status has a positive predictive value (97.7%) for disease phenotype with RS, whereas the absence of RS had a negative predictive value (97.8%) for the mutations.4,17
How SF3B1 mutations contribute to MDS pathogenesis is still unclear. The potential mechanism responsible for iron accumulation in the mitochondria of erythroblasts in MDS characterized by RS is the reduced expression of the ABCB7 gene, which plays essential roles in both mitochondrial iron homeostasis and hematopoiesis.21,22 The down-regulation of ABCB7 has previously been associated with over-expression of FTMT (encoding mitochondrial ferritin) in hematopoietic progenitors.23,24 The mitochondrial ferritin has ferroxidase activity, capturing iron in the ferrous form, depositing as ferric hydroxides after oxidation. The accumulation of this protein in erythroblasts was accompanied by markedly reduced erythroid growth in vitro due to the down-regulation of ABCB7.25 The abnormal mRNA splicing due to mutated SF3B1 might lead to an abnormal ABCB7 protein, deregulating mitochondrial ferritin in the immature red blood cells, thus culminating in the disease phenotype.
Conversely, Visconte et al.26 did not observe any differences in gene expression or exon usage in genes of the mitochondrial pathway, such as ABCB7 and FTMT, in SF3B1-mutant refractory anemia with ring sideroblasts patients. These finds suggest that different biological pathways are involved in the accumulation of iron in hereditary and acquired sideroblastic anemia. Although the authors did not find differences in expression levels of genes relevant to the MDS pathophysiology (such as ASXL1, CBL, EZH1, and RUNX3), these genes were found alternatively spliced in at least one exon, suggesting that SF3B1 mutations are founder mutations that promote subsequent acquisition of genetic abnormalities.
SF3B1 mutations might lead to proteins exhibiting gain- or loss-of-function, which may foster, for instance, clonal expansion and dominance of hematopoietic progenitors carrying the mutation. Malcovati et al.17 showed the assessment of SF3B1 mutant allele burden revealing the presence of a dominant clone with a heterozygous mutation in most of the cases. Moreover, patients carrying this molecular lesion presented a significantly better overall survival compared with those without the mutation, which indicates that SF3B1 mutation analysis is an important predictor of clinical outcomes in these disorders.17
This study is the first to report on SF3B1 gene mutations in Brazilian MDS patients. The frequency of mutations in this splicing gene in patients presenting RS tended to be lower than described in previous studies in European and North American populations. This apparent disagreement may reflect both the heterogeneity of the Brazilian population due to miscegenation and/or the restricted analysis performed in this study, as it focused on hotspot mutations only. MDS is under-diagnosed in Brazil,27 which may also contribute to the differences in mutation frequency observed in the present study. The importance of the molecular analysis in a heterogeneous group of diseases such as MDS is undisputable, especially for improving diagnosis and prognosis.
ConclusionSix heterozygous mutations in the SF3B1 gene were identified in a cohort of 91 Brazilian MDS patients, which were highly correlated with the presence of bone marrow RS, similar to what was previously reported in studies of different populations. This observation highlights the importance of molecular analysis in these diseases besides morphological and cytogenetics analyses of the hematopoietic lineages.
Conflicts of interestThe authors declare no conflicts of interest.
This work was supported by the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) grant 13/08135-2.



