

XIV Eurasian Hematology Oncology Congress
More infoCongenital Factor X (FX) deficiency is an autosomal recessive disorder with variable clinical severity associated with heterozygosis or homozygosis inheritance. Genetic mutations are located on the glutamic acid domain on exon two and the catalytic site of FX on exons 7, 8. Missense mutations of specific patients or families are reported. Severe forms of the disease result from homozygosis or compound heterozygosis genetic mutations. In this case report, we aim to write a rare cause of epistaxis and novel mutations of FX and DMDGH (Dimethylglycine Dehydrogenase) deficiencies, and he and his family are the second with TRAP1-related FEPS1 (familial episodic pain syndrome) in the World.
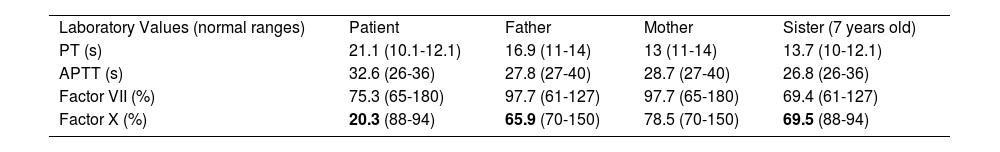
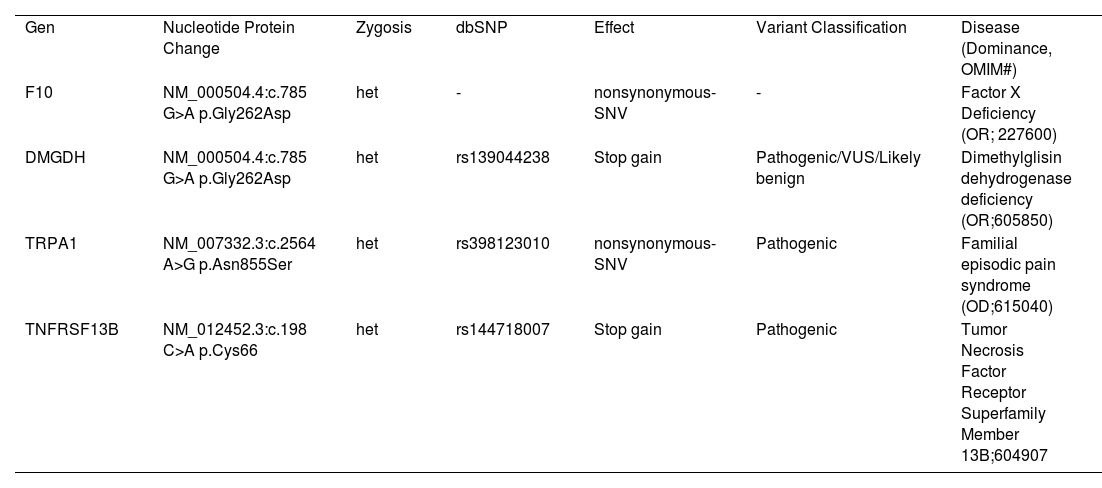
Case reportA 6-year-old boy, born in Turkey, with no known chronic medical condition, was admitted to the pediatric hematology-oncology policlinic with epistaxis lasting for approximately 10 minutes and repeating daily. The family history revealed prolonged bleeding episodes in his father, uncle, aunt, and uncles' sister. The patient denied mucosal bleeding, spontaneous bruising, or prolonged bleeding after dental extraction. The physical examination included; weight: 22 kg (50-75 p), height: 127 cm (>95 p), and regular systemic features. In addition, prolonged PT (21.1s) and normal aPTT levels were found. In his family, prolonged PT was also detected in his father (16.9s) and sister (13.7s). Factor and coagulation levels and their normal ranges consistent with age are given in Table 1. In the clinic exom study, NM_000504.4:c. 785 G>A p.Gly262Asp heterozygosis mutation on the F10 gene has a nonsynonymous_SNV effect, causing Factor X deficiency (Table 1). This mutation is a new change undefined in the Clinvar database with a DANN score of 0.988. According to the ACMG rules (PP3, PM2, PP2), this mutation is pathogenic (Figure 1).
Clinic exom study revealed other mutations (Table 2) associated with the case's clinic features. For example, the patient had a fish odor and muscle tiredness associated with dimethylglycine dehydrogenase deficiency. In addition, the patient suffered from episodic pain syndrome (upper body pain after cold, physical stress, and fasting) and frequent fevers that may be associated with Immunodeficiency and TNFRS13B mutation. Episodic pain syndrome was common in the patient's father's family (uncle, uncle's sister, aunt, grandfather, aunt, and aunt's three children) (Figure 2). However, the diagnosis of immune deficiency is not defined in his family. Family segregation mutation analyses are under study.
ResultsFactor X deficiency is a rare coagulation disorder. The clinic severity differs according to the genetic mutations generally localized to the glutamic domain exon 2 (1). Gokcebay et al. represented an infant with a homogenous FX gene mutation in Exon2 (Gly51Arg) with an FX serum level of 0.03 U/ml. This infant had umbilical cord bleeding and cephalic hematoma and received fresh frozen plasma and activated prothrombin complex concentrate (aPCC). PT (INR) was elevated only(5). Nagaya et al. reported Four heterozygous mutations [p.Gly154Arg, p.Val236Met, p.Gly263Val, and p.Arg387Cys] and a compound heterozygous FX gene mutation (p.Gly406Ser and p.Val424Phe) were identified (6). Another case report showed that a heterozygous nonsense mutation in the F10 gene led to prolonged vaginal bleeding after polypectomy (7).
In neonates, FX levels <10% may cause severe bleeding like CNS, gastrointestinal, hematomas, and hemarthroses. In addition, severe deficiency may cause epistaxis and menorrhagia. The results of the EN-RBD study showed a variable target level of 10% to 20% up to 40% to prevent bleeding. In addition to fresh frozen plasma, Apcc, FIX/FX, and FX concentrates are available to treat FX deficiency. Doses of treatment and schedules differ according to the surgery preparation, prophylaxis, or bleeding. Tranexamic acid is preferred for menorrhagia, nosebleeds, presurgery, and surgery to prevent excessive factor administration (8,9).
Our study showed mild FX deficiency with a nucleotide protein change of NM_000504.4:c.785 G>A p.Gly262Asp, and a novel mutation of FX deficiency. Recurrent epistaxis episodes were controlled with a nasal tampon. We plan to administer tranexamic acid for uncontrolled nasal bleeding. In trauma and surgery, fresh frozen plasma and concentrates are the treatment options.
Familial non-inflammatory pain syndromes (FEPS) are divided into three groups; only one reported family has TRAP1-related FEPS1 syndrome. The predisposing factors for FEPS3 in children are cold, fatigue, hunger, and the rainy season. Pain mainly occurs in the afternoon or at night; paroxysmal pain lasts for tens of minutes, then relieves, and then starts again after a short interval. Unlike FEPS3(cardiac ion channel disease and congenital myotonia), caused by the SCN11A mutation with pain in the distal limbs, TRAP1-related FEPS1 syndrome has pain symptoms in the upper body with autonomic symptoms. Currently, there is no specific drug for treating familial paroxysmal pain syndromes. The primary treatment is to use analgesics, keep warm, and avoid cold environments. Here, we report the second family with TRAP1-related FEPS1 syndrome. The pain is in the upper body, similar to his family (10).
Dimethylglisin dehydrogenase deficiency, as fish odor syndrome, is a likely benign condition with mild muscle involvement (11). We report a novel variant consistent with the clinical situation, a heterozygotic stop gain, NM_013391.3:c.972 G>A p.Trp324 mutation, defined as pathogenic/VUS/likely benign.
ConclusionThis is the first report of F10, DMGDH novel mutations, and the coexistence of TRPA1 (clinic was compatible) and TNFRSF13B with a need for investigation for immunodeficiencies in a child. In conclusion, this is the first patient with two novel mutations for FX and DMDGH deficiencies, and his family is the second with TRAP1-related FEPS1 syndrome in the World.


