
Polycythemia corresponds to abnormally high haemoglobin/ haematocrit in peripheral blood, and identifying the underlying aetiology is crucial for optimizing its management.1 As for its pathophysiology, it may be classified as primary versus secondary and acquired versus congenital. Primary polycythemia is the consequence of mutated progenitor cells and usually presents low erythropoietin (EPO) levels. It may be acquired, as in Polycythemia Vera (PV), when the JAK2 cascade is hyperactivated, or it might be congenital, as in familial congenital polycythemia, resulting from mutations in the EPOR gene. Contrary to primary polycythemia, secondary polycythemia does not result from alterations in the erythroid lineage and when acquired, stems from high EPO levels, which might result from physiological stimuli such as hypoxia, hypoxemic inducing conditions, or tumours with autonomous EPO production. On the other hand, congenital secondary polycythemia arises most frequently from haemoglobin variants that present higher oxygen affinity, or mutations in proteins involved in its homeostasis.2-4 Given the complexity of affiliated conditions, clinical approach to polycythaemia must be exhaustive and multidisciplinary, through a precise balance between clinical practice and aetiological investigation, essential for a more accurate medical practice and therefore a better treatment for the patient.
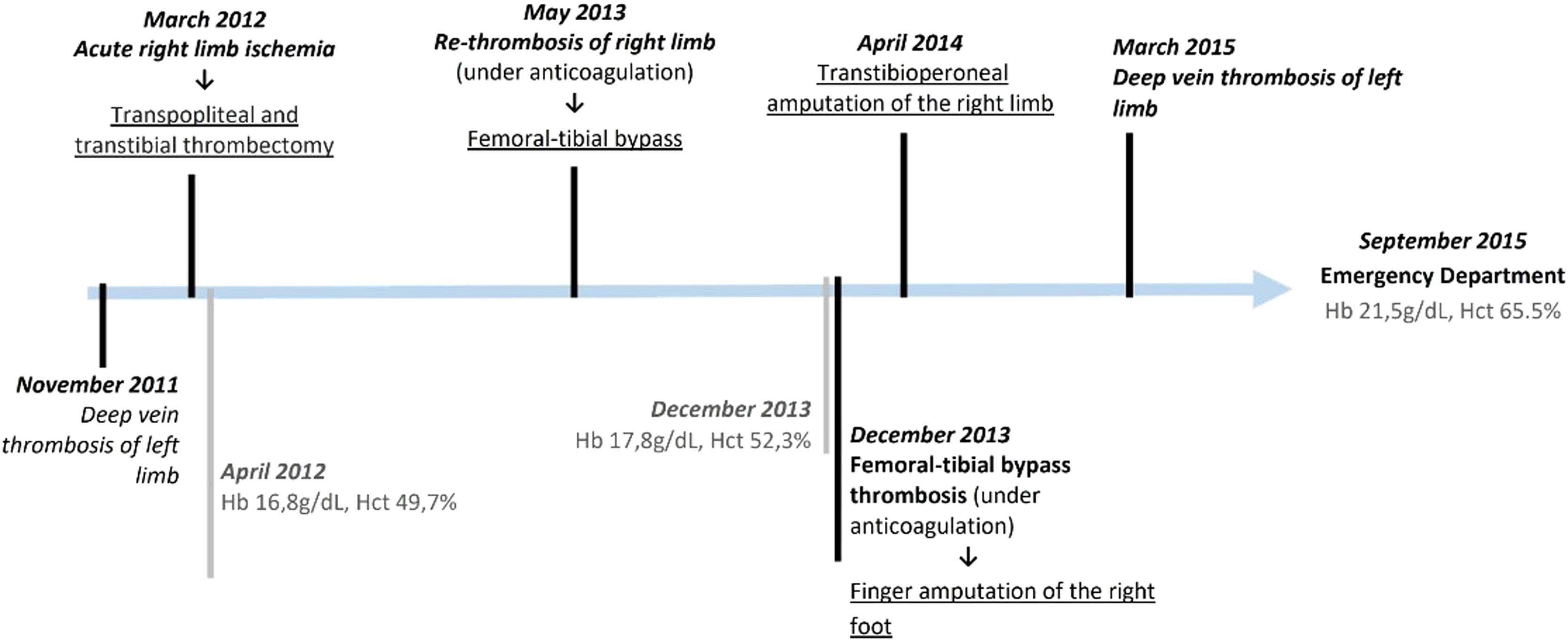
Case reportIn this Article, we present a 49-year-old male with a 20 pack-year history of smoking and severe atherosclerotic vascular disease having presented an episode of deep venous thrombosis of the left popliteal vein (at 45 years old in 2011), and arterial thrombosis of the right lower limb (at 46 years old in 2012). He underwent femoropopliteal bypass surgery with subsequent thrombosis, that culminated in a below-knee amputation (April 2014, Figure 1). He was currently treated with acenocoumarol and acetylsalicylic acid for recurrent thrombotic episodes. The patient did not report relevant haematological history in his immediate family (parents and brother); however, both an uncle and cousin on the paternal side, had a history of thrombotic disease without accompanying polycythemia.

Patient's clinical evolution
Schematic representation of patient's clinical evolution until observation in the Emergency Department. The main thrombotic events and respective therapeutic intervention are highlighted in black; in grey are shown the pertinent laboratorial information available prior to the ER episode.
He was observed in the emergency room (ER) in September 2015 due to coagulation abnormalities. Polycythemia was identified (Hb 21.5 g/dL, Htc 65.5%) without alterations of other haematological lineages, normal reticulocytes (1.04% with WRR=0.5-2.5%) and normal serum erythropoietin (EPO) (5.2mUI/Ml, WRR=2,6-34.0). No morphological changes were found in the peripheral blood smear. The patient was submitted to phlebotomy in the ER and referred for further outpatient study.
In the subsequent outpatient evaluation, previous complete blood counts were reviewed, and discrete polycythemia was found at the time of amputation. As blood counts prior to this surgery could not be obtained, an etiological investigation was conducted. EPO ranged between 3.4 and 16.72mUI/mL after phlebotomy. Sparse megakaryocytes were identified in the sternal bone marrow aspiration, but the bone marrow biopsy was inconclusive showing normal myeloid/erythroid ratio but reduced megakaryocytic lineage with sparse micromegakaryocytes. Karyotype testing came back normal. The abdominal ultrasound did not show hepatosplenomegaly. Non-hematologic etiologies for polycythemia and thrombosis were excluded, such as antiphospholipid syndrome, cardiorespiratory disease (normal arterial blood gas sample and spirometry measured normal; the chest CT detected no structural disease, including emphysema; the polysomnography with no evidence of obstructive sleep apnoea syndrome; the echocardiogram with no relevant alterations) or EPO-secreting tumour (cerebral and abdominopelvic CT). Throughout the study of thrombophilia, only the clinically irrelevant A1298C variant in the MTHFR gene was identified, with normal values of homocysteine.
Given the clinical context, the most likely initial diagnostic hypothesis was Polycythemia Vera (PV), and consequently, the V617F variant in exon 14 of the JAK2 gene (JAK2: c.1849G>T; p.V617F) was investigated although not found. Subsequently, alterations in the CALR and MPL genes were also searched, but also not identified. Variants in exon 12 of JAK2 were also investigated and negative.
Despite the extensive investigation carried out, a conclusive diagnosis was not possible. Nevertheless, the clinical severity and the need for adequate therapy motivated a broadening of the investigation leading to the complete sequencing of the JAK2 gene in DNA extracted from a peripheral blood sample, using a Custom Ampliseq ION S5 next generation sequencing (NGS) approach. This study revealed a heterozygous variant in exon 25 of JAK2 gene (JAK2: c.3323A>G (p.Asn1108Ser)), with an allele frequency of 52% therefore indicative of a germline mutation, later confirmed by DNA extraction from the hair root. This variant was classified as probably pathogenic in the in-silico analysis as it could represent a gain of function in the JAK-STAT pathway. Neither evidence of polycythemia and thrombotic disorder, nor any variant of the JAK2 gene in the sequenced sibling was identified in the immediate family.
Given the uncertainty of the polycythemia presentation timeframe, genetic alterations that cause congenital erythrocytosis were also excluded by a multigene NGS panel performed by Ion AmpliSeq™ (searched genes: BPGM, EGLN1 (PHD2), EGLN2, EGLN3, EPAS1 (HIF2A), EPO, EPOR, HBA1, HBA2, HBB, HIF1A, HIF3A, JAK2, SH2B3 (LNK), VHL). HPLC was performed to discard highaffinity haemoglobin.
Despite the uncertain aetiology, therapeutic phlebotomies were performed aiming for a haematocrit level lower than 45%, and anticoagulation and antiplatelet therapy was kept. Currently, the patient is asymptomatic and without new thrombotic episodes, under anticoagulation and antiaggregation associated to periodic phlebotomies (every 2 months).
DiscussionPolycythemia might reflect multiple conditions, and certain clinical features may guide us to the underlying aetiology. Congenital polycythemias, for instance, occur primarily at younger ages of presentation, in patients with compatible familial records, with polycythemia as the only haematological manifestation and without splenomegaly. On the contrary, the absence of familial history and a diagnosis at an older age favor an acquired aetiology. Nevertheless, the phenotype might be ineffective in determining its aetiology, which makes the genetic study vital.
In this case, given the analytical pattern of polycythemia, impairment of both arterial and venous systems by the thrombotic disease, age at presentation and the absence of cardiopulmonary disease despite previous smoking habits, the most likely diagnosis would be Polycythemia Vera (PV). PV is the most common myeloproliferative neoplasm (MPN) and acquired aetiology of primary polycythemia.5,6 It may be associated with changes in other haematopoietic lineages, but polycythemia is the differentiating factor between different MPN.5 The V617F mutation or mutations in exon 12 of the JAK2 gene are present, correspondingly, in 95% and 3% of PV cases, constituting major criteria for its diagnosis.6 Moreover, cases of PV involving mutations in the CALR gene have also been described.
Despite the status of these mutations in the diagnostic process, none is specific to PV and rare cases of PV, in which no classical molecular marker exists, are documented. In an international review of 1545 patients with PV, Spivak J. was not able to identify a molecular marker in 2% of such patients.5 In these cases, other diagnostic criteria are required, namely a compatible bone biopsy and serum EPO levels below the normal limit.1 However, in this particular case, those were also absent. Then again, clinical reviews of patients with PV show that up to 19% of these as having normal to high EPO levels.6 Therefore, it may be questioned whether the exception to the diagnostic criteria could in fact represent a failure in the diagnostic test itself, as no other cause has been identified to justify the significant polycythemia and thrombotic clinic displayed, despite the extensive investigation carried out.
To avoid underdiagnosis or undertreatment, medical research has used molecular biology and gene sequencing to search for new markers or therapeutic targets. In 2009, Siemiatkowska et. al sequenced the JAK2 gene in 34 JAK2-V617F negative, but suspected MPN patients, and for the first time, identified the N1108S variant of the JAK2 gene in a patient diagnosed with PV.7 In 2016, Cabagnols et. al. sequenced the MPL and JAK2 genes in 17 patients with Essential Thrombocytosis and a triple-negative profile (JAK2V617F, MPL and CALR) identifying, among others, a germline N1108S mutation. The authors theorized that the N1108S mutation could result in a slight gain of function of the JAK2 protein and, in specific individuals, favour the development of MPN.8
In 2019, Benton et. al also sought to clarify the role of JAK2 variants in patient prognosis. They retrospectively studied their frequency in 3376 patients with MPN and/or Acute Myeloid Leukemia (AML) and identified a higher relative frequency of JAK2 variants in patients who progressed from MPN to AML. The N1108S variant, one of the three most frequent in this study, was more common in the group that developed de novo AML and was classified as being likely pathogenic. This variant was also linked to recurrent chromosomal abnormalities and to the TP53 mutation.9 Fernandez et.al. also identified the N1108S variant in a patient with PV who progressed to AML.10 These facts suggest the potential induction of genomic instability of this variant, favouring mutagenesis and leukaemogenesis.11
The significance of this case is not only due to the aggressiveness of the disease's presentation, the exhaustive investigation that it entailed, and the difficulty in devising an adequate therapeutic plan, but also the finding of a rare variant being present. This variant has been described in only two patients with PV, and although its meaning and function are still uncertain, it is theorized that the identification of these variants might represent an advance in the understanding of its repercussions on clinical diagnosis and prognosis, ultimately contributing to a better understanding of the disease and a better approach to our patients. Alongside the aetiological interest in investigating these variants, we also draw attention to the impact that the absence of a diagnosis has on the therapeutic approach, particularly if clinical or analytical stability is not maintained.


