



Acute megakaryoblastic leukemia is characterized by heterogeneous biology and clinical behavior. Immunophenotypic characteristics include the expression of megakaryocytic differentiation markers (e.g. CD41, CD42a, CD42b, CD61) associated with immaturity markers (CD34, CD117, HLA-DR) and myeloid markers (e.g. CD13, CD33) and even with lymphoid cross-lineage markers (e.g. CD7, CD56). Although the diagnostic immunophenotype has already been well described, given the rarity of the disease, its immunophenotypic heterogeneity and post-therapeutic instability, there is no consensus on the combination of monoclonal markers to detect minimal/measurable residual disease (MRD).
Currently, MRD is an important tool for assessing treatment efficacy and prognostic risk. In this study, we evaluated the immunophenotypic profile of MRD in a retrospective cohort of patients diagnosed with acute megakaryoblastic leukemia, to identify which markers, positive or negative, were more stable after treatment and which could be useful for MRD evaluation. The expression profile of each marker was evaluated in sequential MRD samples. In conclusion, the markers evaluated in this study can be combined in an MRD immunophenotypic panel to investigate for megakaryoblastic leukemia. Although this study is retrospective and some data are missing, the information obtained may contribute to prospective studies to validate more specific strategies in the detection of MRD in acute megakaryoblastic leukemia.
Acute megakaryoblastic leukemia (AMKL), classified by the World Health Organization (WHO) 2022 revision as leukemia defined by cell differentiation when the defining genetic abnormalities are unknown, is a rare subtype of acute myeloid leukemia (AML),1 First described by von Boros et al. in 1931, AMKL develops from primitive megakaryoblasts.2,3 AMKL is characterized by the presence of specific antigens of the megakaryocytic lineage in the blast cell membrane as detected by flow cytometry, immunohistochemistry or by the presence of platelet peroxides.4 It is more frequent in children than adults, representing 4–15 % and less than 1 % of AML cases, respectively.5,6 In adults, it tends to occur between the ages of 50 and 60.5,6
For the diagnosis of AMKL, at least 20 % of blasts are required in the bone marrow (BM), of which at least 50 % must be from the megakaryocytic lineage.7 However, morphological identification of megakaryoblasts is not sufficient to diagnose AMKL.8 According to WHO criteria, the diagnosis of megakaryoblastic differentiation must include expression of one or more platelet glycoprotein markers, such as CD41a (glycoprotein IIb), CD42b (glycoprotein lb) or CD61 (glycoprotein IIIa) identified by flow cytometry in blast cells.1
Therefore, the megakaryocytic markers CD41a, CD42b and CD61 are essential for the immunophenotypic characterization of AMKL. The immunophenotypic profile of AMKL also includes the absence of myeloperoxidase expression, the heterogeneous expression of stem cell markers such as CD34, CD117and HLA-DR, the expression of some myeloid markers such as CD13 and CD33 and also CD36, an erythroid and megakaryocytic marker. In addition, the T/NK lymphoid lineage markers CD7 and CD56 are frequently expressed in this leukemia subtype.6,8,9
However, AMKL may has a heterogeneous immunophenotype, with some markers being expressed in only one of the blast subpopulations. More comprehensive studies to define immunophenotypic profiles are limited due to the rarity of the disease.8
AMKL is subdivided in three groups based on cytogenetic and molecular alterations: children with Down Syndrome (DS), children without DS and adults usually without DS.10 Each group has different genetic alterations that are associated with different outcomes.10
Children with DS-AMKL have the uniform presence of trisomy 21 (T21) and GATA1 mutations, which are accompanied by mutations in chromatin regulators such as cohesin subunits and EZH2 or signaling molecules such as JAK/STAT and RAS pathways.10–13 The combination of trisomy 21 and GATA1 is also responsible for the development of transient myeloproliferative disorder or transient abnormal myelopoiesis (TAM) in DS as observed in the first months of life in up to 30 % of newborns with DS.5,10 Most patients with TAM achieve spontaneous resolution within weeks of diagnosis although approximately 10 % to 20 % of patients experience disease progression to AMKL within 3 to 4 years of life.5,14 This progression is associated with the acquisition of additional mutations and clonal expansion.10–13 DS patients older than four years of age rarely have AMKL associated with a GATA1 mutation,15 however, they have an estimated 150 to 500 times greater risk of developing AMKL compared to children without DS.5
In contrast, children with non-DS-AMKL characteristically lack GATA1 mutations.10 The RBM15-MKL1 fusion or t(1;22) (p13.3;q13.1) is pathognomonic of non-DS-AMKL and some recurrent genetic alterations have also been described.5,7,12 The prognosis including mortality for this group is worse than that of children with DS.5,10,12
In adults, the most common molecular abnormalities in AMKL are inv(3)(q21;126), t(9;22)(q34;q11), aberrations of chromosomes 5 and 7, and complex kariotypes.6,11 Molecular changes in adult AMKL may be related to mutations in the cohesin and splicing component genes, as well as TP53 and DNMT3A.10,12
AMKL can also evolve from a myelodysplastic neoplasm (MDS) or a myeloproliferative neoplasm.3
The coexistence of MDS and acute megakaryoblastic leukemia usually causes an aggressive behavior of the disease, which is more prevalent in the elderly, although reports in the literature are rare.3 Cytogenetic abnormalities, including trisomy (8, −7q, −5q) and translocations [t(1;22), t(1;5) and t(8;17)], are found more frequently in adults with AMKL secondary to MDS than in other types of AMKL.3
A presence of t (9;22) indicates transformation from chronic myeloid leukemia.3 AMKL can evolve from essential thrombocythemia with a risk of leukemic transformation varying from 1 to 4 %.16
Adult patients with AMKL tend to have a worse prognosis than pediatric patients with an overall survival of less than one year, even for patients undergoing allogeneic hematopoietic stem cell transplantation (alloHSCT).17
There are few descriptions in the literature about the association of specific immunophenotypic profiles associated with genetic subsets of AMKL, as has been described in other acute leukemias.8,18,19
The persistence of chemo-resistant leukemia cells after the induction of leukemia remission, that is minimal/measurable residual disease (MRD), is an important predictor of relapse and shorter survival in patients with AML.20,21 Due to its prognostic relevance, the information about the MRD status helps clinical decisions about post-remission therapy to prevent leukemia relapse based on the risk of disease recurrence, especially when associated with genetic risk information.22 Therefore, MRD is a predictive biomarker for AML in subsets of patients at different times of treatment, such as after induction and consolidation therapy, and before and after hematopoietic stem cell transplantation (HSCT).23
One of the most applicable methods to monitor MRD in AML is immunophenotyping by multiparametric flow cytometry, through the detection of aberrantly expressed markers in leukemia cells. The ability of current flow cytometers to detect eight or more markers simultaneously enables a more accurate MRD analysis.21 However, this potential can only be achieved if sufficiently informative monoclonal markers are used, together with the application of strictly controlled pre-analytical processes.21
The recommendation of the European LeukemiaNet (ELN) 2021 for MRD assessment in AML includes markers for leukemia-associated immunophenotypes (LAIPs) observed at the time of diagnosis and for aberrant immunophenotypes acquired after treatment (“different from normal”). 21 This enables the detection of early clones and emerging leukemic clones, which may appear and/or disappear during treatment and MRD monitoring.21 Both approaches require experience in recognizing aberrant populations among normal background cell populations, as well as establishing validation strategies for MRD assessment.21
The core markers recommended by the ELN 2021 for assessing MRD in AML are CD34, CD117, CD45, CD33, CD13, CD56, CD7, HLA-DR.21
Given the phenotypic instability of AML after treatment, it is important to recognize the stability and frequency of expression of different markers during therapy. Due to the rarity of AMKL, there is no consensus in the literature about the most relevant markers for the detection of MRD. This study aims to retrospectively evaluate the diagnostic and post-treatment immunophenotypic profiles of AMKL patients to identify potentially useful markers for MRD detection.
Patients and methodsThis study was approved by the institutional ethics committee (CAAE: 60695522.7.0000.5434).
PatientsThe inclusion criterion for patients in the study was the diagnosis of AMKL. Patients lacking information on the immunophenotype of the diagnosis or evaluation of MRD were excluded from the study. Seven patients, aged 1 to 52 years, diagnosed with AMKL according to WHO criteria were included in the study.
Of the included patients, four were female (57 %) and three were male (43 %) (five pediatric (71 %) and two (28 %) adults). Two pediatric patients had Down Syndrome. Six patients were diagnosed with primary AMKL and one with AMKL secondary to MDS.
Six out of the seven patients underwent allogeneic HSCT. Four of the six transplanted patients died: one of leukemia relapse, one of acute pulmonary graft-versus-host disease (GVHD), one of graft loss and one of sepsis. Of the two patients who survived HSCT, one diagnosed with MDS had trisomy 8 and an IHD2 mutation and one child had non-DS-AMKL. One pediatric patient with DS-AMKL was not transplanted and died from treatment complications.
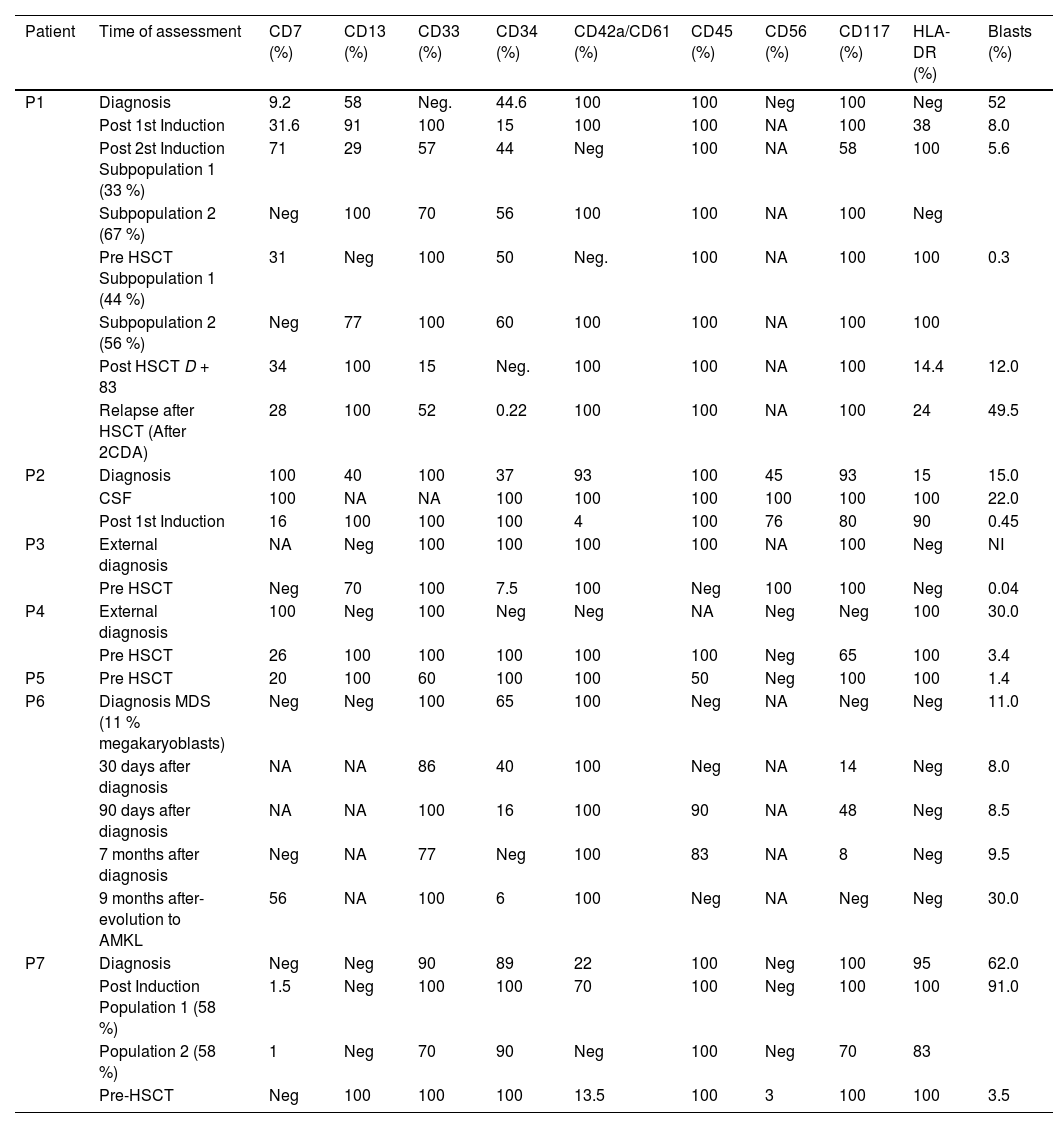
Table 1S shows the characteristics and clinical evolution of these patients.
MethodsData collectionThis retrospective study used the flow cytometry laboratory database and information from the medical records of patients treated with AMKL at the Hospital Amaral Carvalho from January 2012 to December 2022. The diagnostic immunophenotypes were compared with the MRD immunophenotype of 18 samples evaluated at each evaluation moment, such as after induction, consolidation, and pre and post-HSCT. Three patients were not diagnosed at the institution but had information about the initial immunophenotype in their medical records.
Data evaluationDescriptive analysis was performed, comparing the percentages and expression intensities of each marker at the time of diagnosis and post-treatment (MRD) to assess losses, gains or stability of expressions. By definition, the expression of a marker was considered positive when its intensity was above one decade in a fluorescence scatterplot in the FCS file (FCS). In addition, lymphocytes from each sample were used as negative internal controls for the myeloid lineage. Intensities of the marker expressions were defined according to the description in the reports. Any MRD value found was considered for the study, even if it was below the cutoff point considered clinically relevant (0.1 %), as the objective was to assess the stability of monoclonal markers at different times of treatment, regardless of the MRD level. The percentages of samples with the respective expression of each immunophenotypic marker in the MRD evaluations were calculated in relation to the total number of evaluable samples for each marker. In this evaluation, even samples without the expression of some markers at the time of diagnosis, but which were observed after treatment, were included.
ImmunophenotypingThe pre-analytical and analytical protocols were used as previously described, including the AML diagnostic panel.9,23 These standard operating procedures (SOPs) are used daily in our laboratory to ensure the intra-laboratory linearity and reproducibility of the tests.
For MRD assessment, 8-color monoclonal antibody panels were customized according to the initial immunophenotype, but some markers were common for most cases, such as CD7, CD13, CD33, CD34, CD45, CD56, CD117 and HLA-DR. For this reason, they were selected for the study and because they are the same markers recommended by the ELN for MRD in AML.21 Furthermore, two megakaryocytic differentiation markers, CD42a and CD61, were included both conjugated to the same fluorochrome. Both LAIPs and different-from-normal approaches were used for MRD detection in this study. Table 2S shows the specifications of the markers selected for the study.
Samples were acquired in BD FACSCanto II and BD FACSLyric 10 flow cytometers. The number of acquired events ranged from 1 to 5 million per tube. Bulk lysis was used to concentrate the sample and achieve 5 million acquired events per tube.
The software used for the analyses was Infinicyt (Cytognos)™. Figure 1S shows the differentiation between true expression of megakaryocytic markers in blast cells and platelet satellitism. Figure 2S illustrates a gating strategy used for case analysis.
ResultsDuring treatment, almost all the markers studied showed variations in their expression and in the percentage of positive cells.
Five out of the seven cases evaluated had CD7 expression at diagnosis, which was maintained during the evolution, although fluctuations in cell frequency (increases and decreases) and in the intensity of marker expression were observed (Tables 2 and 3S). Two cases did not show CD7 expression at diagnosis, but became positive in MRD evaluations and one of them became negative again in the pre-HSCT sample (Table 3S).
CD13 was present in two cases at diagnosis, but was expressed in six cases in MRD assessments with variations in the intensity of expression (Table 2). In the two initial cases, CD13 showed heterogeneous expression at diagnosis, homogeneous expression during treatment, with expression oscillations and even absence of expression (Table 3S).
Expression percentages of each monoclonal marker in the diagnosis and assessment of MRD at different stages of treatment in patients with AMKL.
MDR: Minimal/measurable Residual Disease; AMKL: Acute Megakaryoblastic Leukemia; HSCT: hematopoietic stem cell transplantation; MDS: myelodisplasic neoplasm; CSF: cerebrospinal fluid; NA: not assessed; NI: not informed; Neg: negative.
CD33 was detected in six cases at diagnosis and in all seven cases studied during the MRD assessments. Variations in the intensity of expression and in percentage of positivity were observed throughout the evolution (Tables 2 and 3S).
CD34 is frequently expressed on AMKL cells.8 In this series CD34 was expressed in all seven studied cases. During evolution, CD34 showed a reduction in the percentage of positive cells and loss of expression in two of the seven studied cases, with stable expression intensity in the remaining cases (Table 3S).
CD45 was evaluated in five cases at diagnosis and was expressed in four of them. CD45 expression was weak and homogeneous, but in two cases it became heterogeneous during treatment. The case without CD45 expression at diagnosis had transient positive expression during treatment. Information on CD45 was lacking in the other two cases as they were not initially diagnosed at our institution, but in the MRD evaluations both were positive (Table 3S).
CD117, a stem cell marker, was positive in 6/7 cases. In the case without expression of the marker, there was a transitory positivity in leukemic cells in the MRD evaluations. It was observed that the expression of this marker was always weak but stable (Tables 2 and 3S).
HLA-DR was the marker with the lowest frequency of expression at diagnosis (3/7 cases) but became positive in two of the four negative cases. Its expression oscillated between heterogeneous and positive in 5/7 cases (Table 3S).
CD56 was studied in 5/7 cases and was expressed in two. In one case there was only one MRD assessment and in the other the expression of this marker remained positive after treatment.
The megakaryocytic lineage-specific markers, CD42a and CD61, were expressed in 6/7 cases evaluated at diagnosis. They were positive in the MRD assessment in all seven cases, showing transient negativity in only one of the seven cases. The combination of CD42a and CD61 had homogeneous expressions at all the time points of treatment in 7/7 cases. In 2/7 cases there was positivity in only one of the blast cell subpopulations (Tables 2 and 3S).
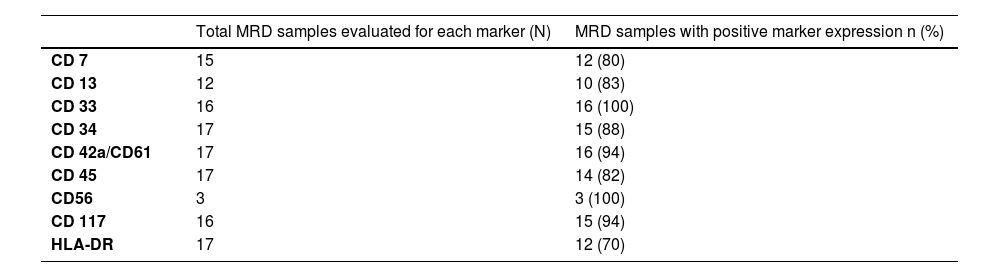
The percentages of samples with the presence of each immunophenotypic marker in the MRD assessments are shown in Table 1.
DiscussionThis study aimed to identify better strategies for the detection of MRD in AMKL. There is little information in the literature with sufficient sensitivity and precision for clinical use that allows the establishment of a standardized protocol to assess MRD in AMKL.
AMKL does not have a reliable molecular marker for monitoring MRD, although a few studies have used GATA1 mutations in non-clinical trial settings.24–26 There are no references on the detection of MRD using other molecular lesions related to AMKL.
Multiparametric flow cytometry is applicable in > 90 % of patients to monitor MRD in AML with adequate sensitivity for use in the clinical practice considering the clinically relevant cutoff of 0.1 %.21 Therefore it is necessary to recognize robust immunophenotypic markers and use appropriate analysis strategies to accurately detect residual leukemia cells. In addition to the appropriate choice of a monoclonal marker combination, rigorous and continuous use of pre-analytical and analytical SOPs are essential to ensure reliable MRD results.27,28
Lack of post-therapeutic phenotypic stability in AML has been described and is expected in MRD assessments.29 Despite the small number of AMKL samples in this study, there were some variations in the percentage of positive cells and expression intensities of the studied markers (CD7, CD13, CD33, CD34, CD45, CD56, CD117 and HLA-DR) during the treatment which did not compromise MRD detection. This indicates the usefulness of these markers, the same markers previously recommended for MRD in AML, in panels targeting MRD of AMKL.21
It should be noted that CD34, CD45, CD117 and HLA-DRusually have variable expressions in AMKL8, and both the absence and positive expression of one or more can contribute to the diagnosis.8 These variations can characterize the asynchrony of expression of these markers and, as seen in this study, are suitable for the detection of MRD.
Although all markers selected in this study were relatively stable after treatment, CD33 and CD117 showed greater stability, demonstrating that they can be used as backbone markers in the MRD panel for AMKL.
CD13, CD7 and CD56 were not expressed in all samples at the time of diagnosis, but showed a high frequency of positivity in samples during treatment and played a role in the detection of residual cells.
Although the use of megakaryocytic lineage markers to detect AMKL MRD seems obvious, there are no descriptions of their stability after therapy. The combination of CD42a and CD61 proved to be quite stable (94 % positivity) in the MRD post-induction and pre- and post-HSCT evaluations in respect to the diagnostic samples. Furthermore, in the two cases with positivity for these two markers in only one subpopulation of blasts at diagnosis, there was persistence of only the population expressing CD42a/CD61, indicating that perhaps this cell population is more resistant to treatment. In conclusion, both markers have high sensitivity for diagnosing megakaryoblasts8 and can be considered reliable for MRD assessments in AMKL.
Therefore, we conclude that the markers selected for this study can be combined into a panel for MRD of AMKL.
It should be noted that there are few reports in the literature on the use of MRD in AMKL to assess response to treatment, but there are no descriptions of the monoclonal markers used in these studies, which would allow comparison with our data. Some reports associate immunophenotypic markers with prognosis30–33, but there are no references on the stability of these markers after treatment.
AMKL is a rare disease with a poor prognosis.2,3,5 An accurate MRD result can provide information about the biological behavior of the disease so that the most appropriate treatment can be offered in each situation, especially for patients with Down Syndrome, who are more susceptible to treatment toxicity.13,14
The limitations of this study was its retrospective nature with some missing data and a small number of cases due to the rarity of the disease. Despite of this, the information obtained here can contribute to prospective and multicentric studies to validate more specific strategies in the detection of MRD in AMKL including the real role of megakaryocytic markers, thereby contributing to the management of these patients.
CRediT authorship contribution statementCarina Maria Pinto: Data curation, Investigation, Writing – original draft. Camila Marques Bertolucci: Methodology, Supervision. Alef Rafael Severino: Methodology, Supervision. Juliana Fernanda dos Santos Tosi: Methodology, Supervision. Maura R V Ikoma-Colturato: Conceptualization, Formal analysis, Writing – review & editing.



{kind=link}
{kind=link}