



Improvements in clinical assessment have occurred since the last published recommendations on the diagnosis and treatment of acute promyelocytic leukemia in 2013. Here, a committee of specialists of the Brazilian Association of Hematology, Hemotherapy and Cellular Therapy presents a comprehensive review on the current knowledge, focusing on the advances in diagnosis, risk assessment, and frontline and salvage therapy. The concept of urgent diagnosis is explored as well as the management of critical situations such as coagulopathy and differentiation syndrome. Recent adjustments in risk stratification based on white blood cell counts only are presented together with the incorporation of chemo-free regimens for non-high-risk patients. Special conditions such as acute promyelocytic leukemia in children, the elderly and pregnant women are discussed. Finally, acute promyelocytic leukemia is presented as a highly curable disease because of the real possibility of targeted therapy towards differentiation, and, paradoxically, as a serious and urgent condition that deserves prompt recognition and management to avoid early mortality.
Acute promyelocytic leukemia (APL), a distinct subtype of acute myeloid leukemia (AML), is characterized by a specific chromosomal translocation involving the retinoic acid receptor alpha (RAR-α) gene on chromosome 17 and the presence of abnormal promyelocytes in the bone marrow.1,2 APL is usually associated with a complex coagulopathy which may cause severe bleeding, with intracerebral and pulmonary hemorrhages being the main causes of death both prior to and at the beginning of therapy.
The fusion protein PML::RARA forms complexes with diverse classes of proteins, enhancing its association with corepressors and functioning as a repressor that remains unaffected by physiological concentrations of all-trans retinoic acid (ATRA; tretinoin). Consequently, it inhibits the transcription of multiple genes involved in myeloid differentiation by altered recruitment of histone deacetylases. ATRA triggers the proteasome-mediated degradation of PML::RARA and transcriptional activation, leading to differentiation. Additionally, arsenic trioxide (ATO), more recently introduced in APL therapy, induces post-transcriptional modifications in the second B2 domain of PML, resulting in the restoration of nuclear bodies and degradation of PML::RARA.3
Epidemiologically, APL accounts for approximately 5–10 % of all AML cases, with a slight predilection for adults over children and the elderly. The incidence rates vary across different populations, with the highest reported rates in Latin America (20–25 % of AML cases), followed by Asia and Europe. Cases of therapy-related APL are uncommon, with a relative frequency close to 5 % and a markedly shorter latency time to diagnosis and similar prognosis when compared to those with de novo APL. APL shows no gender predilection and affects individuals across a wide age range, although it is more common in middle-aged adults.4,5
Incorporating ATRA in the treatment of APL has had a transformative impact on the management and prognosis of this disease. ATRA represents a noteworthy advancement in cancer treatment, serving as a prime example of targeted therapy at the molecular level. The addition of ATO, known for its potent biological activity in APL, has further improved clinical outcomes.6,7 Multicenter clinical trials have demonstrated survival rates exceeding 70 %. A successful cure of APL patients depends not only on the effective utilization of combination therapies that incorporate differentiating and traditional cytotoxic agents but also on comprehensive supportive care measures tailored to the unique biology of the disease and the potential complications associated with targeted molecular therapies.
Additionally, it is crucial to recognize the diagnostic suspicion of APL as a medical emergency since it implies immediate initiation of ATRA therapy, prompt genetic diagnosis, and interventions to manage the coagulopathy.
From its first description in 1957 as a fatal acute hemorrhagic disease until the present day when molecularly targeted therapy is available, APL has gone from highly fatal to a highly curable leukemia especially due to the incorporation of target and non-chemotherapy-based treatment. There are excellent reviews and clinical guidelines addressing the management of APL.6–9 In Brazil, the last published recommendations on APL management date from 2013, when the association of ATRA and ATO was not a current practice.10 Therefore, the creation of a Brazilian consensus is particularly needed because:
- 1)
APL is a peculiar subtype of AML that requires specific treatment;
- 2)
in Brazil, APL is treated in specialized centers and non-specialized centers, and the incorporation of ATO is relatively new; and
- 3)
in addition to the recent remarkable outcomes achieved by current treatment of APL, the critical aspects of APL management must not be underestimated.
In this article, we describe the principles of diagnosis and management of APL, considering the Brazilian healthcare setting.
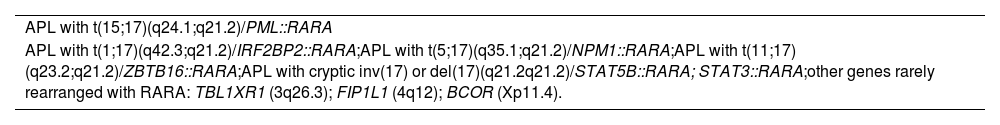
Acute promyelocytic leukemia diagnosisCurrent definition of acute promyelocytic leukemiaAccording to the recent International Consensus Classification on Myeloid Neoplasms, similar to all recurrent genetic abnormalities in AML [except for AML with t(9;22)(q34.1;q11.2)/BCR::ABL1], APL diagnostic criteria are currently recognized with the presence of at least 10 % of blasts in the bone marrow or blood plus specific genetic abnormalities.1,11 Two distinct subtypes [APL with classic t(15;17)(q24.1;q21.2)/PML::RARA and APL with other recurring RARA translocations] are included in AML with recurrent genetic abnormalities (Table 1).
APL with recurrent genetic abnormalities (requiring >10 % blasts in BM or PB).
The genetic alteration is directly related to impairment of myeloid maturation at the promyelocyte stage, often resulting in the accumulation of blasts with the appearance of abnormal promyelocytes in the bone marrow and/or peripheral blood, along with consequent cytopenias as well as a typical coagulopathy that manifests as a high risk of bleeding and requires rapid diagnosis and treatment to prevent early fatality.
Initial clinical and laboratorial workup of patients with suspected acute promyelocytic leukemiaThe initial workup for the diagnosis of APL requires clinical information, medical history and physical examination combined with morphology of the cells, immunophenotyping, karyotype analysis and molecular genetic testing, as reviewed below.1,12,13
Clinical complaintsPatients with APL usually complain of fatigue or weakness due to anemia and fever caused by infection related to neutropenia and bleeding. The medical history includes patient´s age, sex, ethnicity, history of any previous hematologic disorder or predisposing condition, prior malignancy, exposure to radiotherapy, chemotherapy or immunotherapy or toxic substances; and confounding factors, such as recent growth factor therapy, or medications that may mimic acute leukemia. Additionally, family history of any hematologic disorder or malignancy is important information for an occasional diagnostic qualifier. Rare cases have extramedullary involvement.
Complete blood count and morphology of cellsThe starting point for the workup is the complete blood count and the inspection of a peripheral blood smear. Most patients (70 %) present pancytopenia with cells resembling abnormal promyelocytes with eccentric and bilobed nucleus, folded contour and prominent nucleolus. The cell cytoplasm presents irregular azurophilic granules and Auer rods, sometimes containing bundles of Auer rods, called Faggot cells (hypergranular variant). Some cases have leukocytosis instead of leukopenia, and the microgranular variant. The microgranular or hypogranular variant shows paucity or absence of granules and bilobed nuclei. Classically, the white blood cell (WBC) count at presentation is a prognostic factor, differentiating patients into high- (WBC count >10×103/µL) versus intermediate- and low-risk groups, further stratified according to platelet count (WBC count <10 × 103/µL and platelet count above or below 40 × 103/µL). More recently, only the WBC is considered for risk-based treatment decisions. Anemia and thrombocytopenia are frequent and if the values of hemoglobin and platelets are dangerously low, immediate transfusions are indicated.
Bone marrow aspirationA fresh bone marrow (BM) aspiration is necessary for morphologic evaluation, immunophenotyping and for G-banding karyotype. Marrow aspirate smears stained with Giemsa, Wright-Giemsa, May-Grunwald-Giemsa, or others, are optimal for blast enumeration, differential cell counts and morphology analysis. These smears usually show a hypercellular marrow and confirm the presence of atypical promyelocytes with the morphology described above. The number of blasts/promyelocytes are >20 % in most APL cases, however diagnosis of APL may be concluded in the presence of t(15;17)(q24.1;q21.2) or PML::RARA even when blasts/promyelocytes <10 %.2
ImmunophenotypingA multiparametric flow cytometry immunophenotyping panel should be sufficient to distinguish APL from other AML subtypes. Atypical promyelocytes show positive expression of CD13, CD33 and CD117; and low or negative CD34, CD11b, and HLA-DR. Some AML cases may mimic APL but can usually be distinguished by the CD2, CD13, CD34, CD64, and MPO expressions. One of the most challenging differential diagnoses has to be made with AML with the NPM1 mutation that resembles APL because of HLA-DR-negative blasts; therefore, all leukemia cases lacking HLA-DR expression need molecular exclusion of the PML::RARA rearrangement.
KaryotypeG-banding karyotype is mandatory and allows the diagnosis of APL in most cases. The BM sample must be collected in heparin and sent immediately to the laboratory. Karyotype frequently shows the balanced translocation t(15;17)(q24.1;q21.2), corresponding to the molecular PML (promyelocytic) and RARA (retinoic acid receptor alpha) gene fusion. Additional chromosomal aberrations are seen in more than 40 % of cases, such as trisomy of chromosome 8, deletion of 7q, among others, which do not affect the prognosis.14 Rarely, there may be variant translocations or fusions involving the RARA gene, such as t(5;17)(q35;q12–21) or NPM1::RARA, t(11;17)(q23;q21) or ZBTB16 (former PLZF)::RARA, t(11;17)(q13;q21) or NUMA::RARA, del(17) or inv(17)(q21.2q21.2) or STAB5::RARA or STAT3::RARA, t(1;17)(q42.3;q21) or IRF2BP2::RARA, besides TBL1XR1(3q26), FIP1L1 (4q12), BCOR (Xp11.4), all classified as APL with RARA rearrangements.1 Not all of these fusions are detected by conventional cytogenetics but by molecular methods. Karyotype is normal in <10 % of APL cases due to cryptic or masked translocations and in these situations FISH, real-time quantitative polymerase chain reaction (RT-qPCR) and/or genomic testing may complement the investigation.15
Fluorescent in situ hybridizationThe utility of fluorescent in situ hybridization (FISH) should be considered in the context of the case. FISH for the PML::RARA rearrangement may be carried out with peripheral blood or BM samples and offers detection of the PML::RARA translocation within hours (< 24 h). Thus, it could be ordered when in need of a fast diagnosis, or when cytogenetic analysis is suboptimal due to poor chromosome morphology or has insufficient growth. Rare cases may show variant fusions as well.
Molecular genetic testingReal time quantitative polymerase chain reactionReal time quantitative polymerase chain reaction (RT-qPCR) is the gold standard method for diagnosis and treatment monitoring. The test allows the rapid amplification of the specific segment of cDNA and the quantification of its generation in the qPCR assay. Depending on the breakpoint location on chromosome 15, three isoforms can be detected: long (bcr1, intron 6), variant (bcr2, exon 6) or short (bcr3, exon 3). Sporadic cases may present atypical isoforms not detected by conventional primers. In these situations, it may be necessary to sequence the fusion gene. At diagnosis, this test may be done using BM or peripheral blood samples, but for monitoring most protocols suggest using BM samples only.
Next generation sequencingNext generation sequencing (NGS) or massive, parallel, high throughput sequencing is a revolutionary method of DNA and RNA sequencing. The workflow consists of library preparation, sequencing and data analysis. The clinical use of NGS has increased in the last decade. It has shown a mutational landscape in APL different from other AMLs. The PML::RARA abnormality is not able to trigger the leukemic phenotype alone. Additional somatic mutations occur in other genes such as the FLT3, WT1, NRAS and KRAS genes. Around 70 % of patients harbor one to two somatic mutations in addition to PML::RARA. Mutations in genes associated with signaling pathways (FLT3, N/KRAs), tumor suppression (WT1), chromatin organization (ARID1B, ARID1A), oncogenes (SALL4, MD12, NSD1), DNA methylation (DNMT3A, IDH1/2, TET2), NPM1, and epigenetic regulation (ASXL1) are described.3 The FLT3 internal tandem duplication (ITD) and tyrosine kinase domain (TKD) mutation, as D835, are the most frequent co-occurring events but with controversial prognostic implications and it is not recommended to change therapy in these cases. However, at relapse APL patients frequently show additional mutations, mostly acquired during the course of the disease. Point mutations in the PML:RARA fusion gene may also appear in relapse. In this scenario, NGS is being recommended for refractory and relapsed cases.
The procedures and tests required to diagnose AML, including APL, are detailed in Table 2. All options described in the genetic analysis in Table 2 are equally specific to diagnose APL.6,7 Conventional karyotype is less efficient than the other techniques since it requires enough metaphases for analysis. The results of FISH and immunostaining with anti-PML monoclonal antibodies are usually fast and useful to diagnose patients with APL that lack the conventional PML::RARA fusion. Nevertheless, it is essential to note that these techniques should not be considered substitutes for reverse transcription polymerase chain reaction (RT-PCR), as this technique enables the identification of the specific RARA rearrangements and serves as the gold standard for evaluating minimal residual disease.6,7
Tests to establish APL diagnosis.
| Medical historyDemographics and medical recordFamily historyAnalysis of comorbidities |
| MorphologyPeripheral blood count and differential countaBone marrow aspirate and smearbMyeloperoxidase (cytochemistry)c |
| Flow cytometryBone marrow aspirate and smeard |
| Genetic analysisConventional karyotype eFluorescent in situ hybridizationfRT-PCR of PML::RARAImmunofluorescence with anti-PML monoclonal antibodyg |
| Additional testsPerformance statusGeriatric assessmenthCoagulation tests and biochemistryiScreening for hepatitis B, and C, HIVß-HCG testjLumbar puncturekReproductive counselingl |
Can increase the accuracy of morphological analysis. Frequently, the APL blasts show SSC high, CD45 int, CD34-, HLA-DR-, CD117+/-, CD13 +, CD33++, CD15-, CD11b-. The morphological hypogranular variant commonly expresses CD2.
At least 20 bone marrow metaphases should be analyzed to define abnormal karyotype, including RARA rearrangements.
If analyzable metaphases are not available, fluorescence in-situ hybridization (FISH) serves as an alternative approach for detecting genetic abnormalities.
Highly specific for the presence of PML::RARA fusion, the blast cells demonstrate > 30 dots in the nuclei (microspeckled staining pattern). The test is also positive for other RARA rearrangements.
Includes: glucose, creatinine, urea, sodium, potassium, phosphorous, magnesium, calcium, uric acid, aspartate aminotransferase, alanine aminotransferase, alkaline phosphatase, lactate dehydrogenase, bilirubin (total and fractions), prothrombin time (PT), activated partial thromboplastin time (aPTT), d-dimers, fibrinogen.
Patients with APL are at risk of disseminated intravascular coagulation with fibrin degradation products, high d-dimer level and low fibrinogen. Coagulation tests are recommended, including prothrombin time (PT), activated partial thromboplastin time (aPTT), fibrinogen, and D-dimers. Coagulopathy is consistent with elevated partial thromboplastin time, PT, D-dimers and hypofibrinogenemia besides thrombocytopenia. As fibrinogen is an acute phase protein, it may take some time to decrease, so the other parameters may already be low while fibrinogen is still normal. Nevertheless, decreased levels of fibrinogen and prolonged PT represent the most reliable parameters for predicting bleeding.16–19
Acute promyelocytic leukemia treatmentSupportive measuresAll-trans retinoic acid initiation for suspected acute promyelocytic leukemiaATRA should be initiated immediately after the minimum suspicion of APL diagnosis to counteract the disease-associated risk of bleeding. ATRA does not have a deleterious effect in treating other AML subtypes and its early introduction may prevent death by hemorrhage since it improves the manifestations of APL coagulopathy. Based on this, treatment with ATRA should be initiated without delay for suspected APL patients and thus should not wait for molecular APL diagnosis. If molecular confirmation is obtained, treatment including ATRA is continued. In contrast, if typical APL with t(15;17)/PML::RARA is excluded, therapy is adjusted to classical AML protocols.
Support interventions to mitigate acute promyelocytic leukemia coagulopathyApproximately 5 % of APL patients are ineligible for treatment due to fatal or life-threatening hemorrhages that occur prior to the initiation of treatment.20 On this basis, the European LeukemiaNet recommends the use of cryoprecipitate, fresh frozen plasma, and platelets in order to counteract the coagulopathy. The goal is to sustain fibrinogen above 150 mg/dL, platelet count >50×103/µL, and PT and aPTT below 1.5. Replacement therapy should be continued throughout the induction therapy phase until all clinical and laboratory indications of coagulopathy have been resolved.6,7
The prophylactic use of heparin, tranexamic acid, and epsilon-aminocaproic acid is not recommended.
Invasive procedures, such as central venous catheterization and lumbar puncture, should be avoided during the induction phase because of the high hemorrhagic risk during the initial phase of APL treatment.6,7
Management of hyperleukocytosis (white blood cell count ≥10 × 103/µL)APL patients usually present with pancytopenia. However, sometimes they may present with leukocytosis (defined here as a WBC count ≥10 × 103/µL) or an increase in the WBC count may occur during the induction phase of treatment with ATRA and/or ATO. In order to control hyperleukocytosis at presentation, cytoreduction using chemotherapy with idarubicin or daunorubicin should be started promptly in association to ATRA and ATO. Hydroxyurea (Hu) may be used for patients who present or develop leukocytosis after the initiation of therapy with ATRA and ATO to keep the WBC count below 10 × 103/µL. The following recommendation applies as a dosing guide: WBC count 10–50 × 103/µL, Hu 500 mg 4 times a day and WBC count >50 × 103/µL, Hu 1000 mg 4 times a day. Leukapheresis should be avoided because of the risk of fatal hemorrhage.
Central nervous system prophylaxisThe most common site of extramedullary disease and relapse in APL is the central nervous system (CNS).21,22 Patients with neurologic symptoms should be investigated for CNS involvement, although lumbar puncture should be avoided in the early phases of coagulopathy, if possible. There is an increased incidence of CNS involvement in patients with hyperleukocytosis23 however, whether CNS prophylaxis in this group should be applied remains controversial; it is not mandatory in current practice.7 In general, it is advisable to postpone CNS prophylaxis until achieving complete remission (CR) because lumbar puncture can be hazardous at diagnosis and while coagulopathy risk persists.
Treatment approachesBased on the trials of the PETHEMA (Spanish Cooperative Group - Programa Español de Tratamientos en Hematología) and GIMEMA (Italian cooperative group - Gruppo Italiano Malattie Ematologiche dell'Adulto) groups, after remission induction, consolidation treatment has classically been adapted according to risk categories (low-, intermediate-, or high-risk).24 Low- and intermediate-risk categories (presenting platelet counts greater and lower than 40 × 103/μL, respectively) are characterized by WBC counts ≤10 × 103/μL at presentation. Patients presenting with WBC counts >10 × 103/μL are considered to be high-risk. More recently, the prognostic value of the WBC count at diagnosis has been recognized as the most important as it is associated not only with overall survival (OS) and relapse-free survival but also with rates of death and life-threatening complications before or during induction. Therefore, APL is currently stratified into two categories: high-risk (WBC count ≥10 × 103/μL) and low-risk (WBC count <10 × 103/μL), regardless of the platelet count.
Although differentiation therapy with ATRA and ATO is the rule for typical APL with t(15;17)/PML::RARA, in cases of rare RARA translocations, the sensitivity to those agents varies according to RARA rearrangements. For example, cases with t(11;17)/ZBTB16::RARA or der(17)/STAT5b::RARA are poorly responsive to ATRA and ATO.
Non-high-risk patients (white blood cell count <10 × 103/μL)All-trans retinoic acid plus chemotherapyATRA is the mainstream drug in the treatment of APL since its first successful use in a refractory Chinese case in 1985. At that time, the first results that demonstrated CR in all patients treated with ATRA were disseminated.25 Despite the benefit of complete response, ATRA monotherapy was insufficient to promote long-term remission. Subsequently, APL treatment was improved by combining cytotoxic chemotherapy with ATRA.26–29
Thus, front-line treatment consists of ATRA and anthracycline-based chemotherapy.
Sanz et al. compared the treatment outcomes of 350 patients enrolled in the PETHEMA/HOVON LPA2005 with 175 in the Latin-American IC-APL 2005 trials.30–32 Treatment in the two protocols was identical except for the replacement of idarubicin by daunorubicin due to its greater availability and lower cost in the participating countries. Briefly, induction therapy in the PETHEMA trial consisted of oral ATRA 45 mg/m2/day until morphologic CR and intravenous idarubicin 12 mg/m2 on days 2, 4, 6 and 8. The fourth idarubicin dose given on day 8 was omitted in over 70-year-old patients. For under 20-year-old patients, the ATRA dose was adjusted to 25 mg/m2. Patients in CR received three monthly risk-adapted consolidation cycles with ATRA (45 mg/m2/day for 15 days) and chemotherapy according to previously defined risk categories. For low-risk patients, the first cycle consisted of idarubicin (5 mg/m2/day for 4 days), the second of mitoxantrone (10 mg/m2/day for 3 days) and the third of idarubicin (12 mg/m2/day for 1 day). Intermediate-risk patients received a reinforced dose of idarubicin in the first cycle (7 mg/m2/day) and third cycle (12 mg/m2/day for 2 days). For patients who achieved CR after the third course of consolidation maintenance therapy with oral mercaptopurine (50 mg/m2/day), intramuscular or oral methotrexate (15 mg/m2/week) and oral ATRA (45 mg/m2/day for 15 days every 3 months) was given over 2 years. Central nervous system prophylaxis was not given.
The PETHEMA/HOVON cohort demonstrated a significantly higher CR rate of 94 % than the IC-APL cohortʼs 85 % (p-value = 0.002). The distribution of causes of induction failure and the time required to achieve CR were similar in both cohorts. Patients who achieved CR had comparable cumulative incidence of relapse and disease-free survival (DFS) rates in both cohorts. However, the IC-APL cohort showed lower OS and event-free (EFS) survival rates, primarily due to a higher death rate during induction and consolidation therapy. This study indicates that daunorubicin and idarubicin exhibit comparable antileukemic efficacy.30
All-trans retinoic acid plus arsenic trioxideThe combination of ATO and ATRA is currently the standard of care for non-high-risk patients. Two randomized trials confirmed the superiority of ATO plus ATRA over ATRA plus chemotherapy.
The initial trial was a non-inferiority study, conducted by the Italian cooperative group Gruppo Italiano Malattie Ematologiche dell'Adulto (GIMEMA) in collaboration with the German-Austrian AML Study Group (AMLSG) and Study Alliance Leukemia (SAL) cooperative groups, that compared the effectiveness of ATRA plus ATO versus ATRA plus chemotherapy in patients with low-to-intermediate risk APL (WBC count <10 × 103/μL). Patients were randomly assigned to one of two treatment groups: 1) ATRA plus ATO for induction and consolidation therapy, or 2) standard ATRA plus idarubicin induction therapy plus three cycles of consolidation therapy with ATRA plus chemotherapy, along with maintenance therapy using low-dose chemotherapy and ATRA. The results demonstrated that ATO plus ATRA, without chemotherapy, was not inferior and possibly superior to the ATRA plus chemotherapy regimen in terms of EFS and OS. ATO plus ATRA showed less myelosuppression and fewer infections, but it led to more frequent elevations in liver enzymes and QTc prolongation. The update of this trial, analyzing an extended series of patients with a median follow-up of 40.6 months, revealed that the EFS and OS benefits of ATRA plus ATO continued to improve over time. The ATO plus ATRA group exhibited a significantly lower cumulative incidence of relapse and higher OS and EFS, providing further evidence of the superior efficacy of this treatment.
A subsequent trial conducted by the National Cancer Research Institute (NCRI) cooperative group compared ATRA plus chemotherapy with ATRA plus ATO, employing an attenuated ATO schedule in newly diagnosed APL patients. This study included all APL risk groups and demonstrated significantly better relapse-free survival, but no benefit in OS was observed.31,34
A real-world evidence study looked for secondary malignancies after treatment of APL patients with ATRA plus chemotherapy or ATRA plus ATO. From 160 patients in the study, 54 were treated with ATRA plus idarubicin and 106 were treated with ATRA plus ATO. The authors identified 11 cases of secondary cancers, nine of them were induced with ATRA plus Idarubicin and two received ATRA plus ATO. All cases were diagnosed at least one year after APL diagnosis (16–125 months). Among the patients treated with chemotherapy plus ATRA, the authors found the following secondary malignancies: three patients with myelodysplastic syndrome (MDS), two patients with breast cancer, one patient with vulvar cancer, one patient with prostate cancer, one patient with colon cancer and one patient with soft tissue sarcoma. Of the two patients who developed secondary malignancies after receiving ATRA plus ATO, one developed melanoma of the skin and the other developed pancreatic cancer. The authors concluded that ATRA plus ATO did not increase the risk of secondary malignancies.35
Based on the long-term follow-up of the two randomized trials, which show better outcomes and real world evidence and no increased risk of secondary cancer, the combination of ATRA and ATO is the standard of care in non-high-risk APL patients since it may spare patients from using chemotherapeutic drugs. In nations where ATRA plus chemotherapy is more cost-effective than ATRA plus ATO, the conventional approach of combining ATRA with chemotherapy remains an option.7
High-risk patients (white blood cell count ≥10 × 103/µL)High-risk patients may be treated with ATRA plus chemotherapy. In the two aforementioned trials (PETHEMA/HOVON LPA2005 and IC-APL), high-risk patients received the same doses of idarubicin as low-risk patients but combined with cytarabine in the first (1000 mg/m2/day for 4 days) and third cycles (150 mg/m2/8 h for 4 doses), as well as 5 days of mitoxantrone instead of 3 in the second cycle. High-risk over 60-year-old patients did not receive cytarabine and were treated as intermediate-risk patients. The PETHEMA/HOVON LPA2005 trial reported a CR rate of 83 % for high-risk patients and cumulative relapse and OS rates at 3 years of 11 % and 79 %, respectively.31
More recently, strategies based on ATRA plus ATO have been evaluated for high-risk patients. Nevertheless, additional drugs are added to the basic backbone evaluated for non-high-risk patients. Two strategies for high-risk patients have been tested: the combination of ATRA, ATO, and cytoreductive therapy, including gemtuzumab ozogamicin (GO); or the combination of ATRA and chemotherapy.7
Two trials reported the results of the ATRA, ATO, and GO combination in newly APL patients. In the NRCI AML17 trial, high-risk APL patients received 6 mg/m2 of GO on Day 134 and in the MD Anderson Cancer Center Trial, high-risk patients received 9 mg/m2 of GO on Day 1.36 Both trials showed high EFS and OS rates. However, the results reported in these studies did not show significant improvement compared to those reported with ATRA and chemotherapy.37–42
The Australasian Leukaemia and Lymphoma Group (ALLG) compared a historical ATRA-chemotherapy (APML3) protocol with a prospective cohort treated with an induction therapy consisting of ATRA, ATO, and idarubicin (6–12 mg/m2, adjusted for age) followed by two cycles of consolidation with ATRA and ATO, and two years of maintenance with ATRA, 6-mercaptopurine, and methotrexate. After five years of follow-up, the OS was 94 %, and DFS was 95 %. When compared with APML3 data, the hazard ratios were as follows: 0.23 [95 % confidence interval (95 % CI): 0.08–0.64; p-value = 0.002] for freedom from relapse, 0.21 (95 % CI: 0.07–0.59; p-value = 0.001) for DFS, 0.34 (95 % CI: 0.16–0.69; p-value = 0.002) for EFS, and 0.35 (95 % CI: 0.14–0.91; p-value = 0.02) for OS.43 Taken together these findings indicate that ATRA plus ATO and low dose idarubicin is effective in low-risk APL as well as in high-risk patients.
The Shangai group recently published the results of the APL15 trial. In this non-inferiority multicenter trial, 128 patients were randomly assigned to two groups. One group received ATRA plus ATO for induction, consolidation, and maintenance, while the other group received ATRA plus ATO in combination with chemotherapy for induction, followed by three cycles of consolidation therapy and maintenance therapy with ATRA plus ATO. The 2-year DFS and EFS rates in the non-chemotherapy and chemotherapy groups in all-risk patients were 98 % versus 97 % (p-value = 0.62), and 95 % versus 92 % (p-value = 0.39), respectively. In high-risk patients, DFS and EFS were 94 % versus 87 % (p-value = 0.52), and 85 % versus 78 % (p-value = 0.44), respectively. This study demonstrated that ATRA plus ATO had the same efficacy as the ATRA plus ATO plus chemotherapy in the treatment of patients with all-risk APL.44
The lack of large, randomized trials precludes the recommendation of the best therapy combination for high-risk APL patients. Both ATRA plus chemotherapy and ATRA plus ATO and low-dose chemotherapy seem to work well and may be used based upon the availability of drugs and specificity of the patient. When available, patients should be included in prospective trials.
Maintenance after consolidationThe impact and, consequently, the indication for maintenance in the treatment of APL depend fundamentally on two aspects: the combination of drugs used in the initial phase of treatment (ATRA plus chemotherapy versus ATRA plus ATO with or without chemotherapy) and the patient's risk stratification at diagnosis (high-risk versus Intermediate-/low-risk).
After establishing the role of ATRA combined with chemotherapy, the I0129 study from the North American Intergroup Protocol randomized patients to receive maintenance with ATRA or observation after the initial phases. The study demonstrated an advantage in terms of OS and DFS for the subgroup that received maintenance.45 Subsequently, many other cooperative trials were designed to assess the impact of maintenance after initial treatment with ATRA plus chemotherapy. These trials used different strategies: continuous versus intermittent ATRA (15 days of use every 3 months); low-dose continuous chemotherapy (6-mercaptopurine 90 mg/m2/q.d. plus methotrexate 15 mg/m2 q.wk) with or without ATRA or no maintenance. The vast majority of the trials were able to demonstrate the role of maintenance in reducing relapse rates and a trend towards a benefit in terms of OS, especially for the association of intermittent ATRA plus continuous chemotherapy.40,46 The APL 93 study randomized 576 newly diagnosed patients to different maintenance strategies, and after a 10-year follow-up, it demonstrated a significant reduction in the relapse rate when comparing the subgroup without maintenance (43.2 %) to those who received maintenance combining intermittent ATRA plus continuous chemotherapy (13.4 %; p-value <0.001). Another important finding was the demonstration that maintenance was especially beneficial for patients with a WBC count >5 × 103/µL at diagnosis, suggesting that the results of treatment may be improved with the addition of maintenance in high-risk patients.47
On the other hand, some other trials questioned these findings, especially for those patients who achieved complete molecular response after consolidation phases.48 A meta-analysis published in 2013 assessed the role of maintenance in the treatment of APL in the first CR. The systematic review included ten randomized studies with 2072 patients, focusing on three main comparisons: any type of maintenance versus observation; ATRA-based maintenance versus non-ATRA maintenance, and maintenance with ATRA alone versus ATRA combined with chemotherapy. Despite limitations related to the clinical heterogeneity of the studies, particularly regarding the type of maintenance, the authors concluded that there was no significant difference in OS between the strategies. However, maintenance had a favorable impact on DFS, and among the strategies, the combination of ATRA plus chemotherapy seemed to be superior to the use of ATRA alone.49
In the post-ATO era for the initial treatment of APL, maintenance seems to have lost its role, mainly in low- and intermediate-risk groups, as evidenced by the APL0406 and AML17 trials. Both randomized a significant number of patients and did not include maintenance in the treatment strategies. Nevertheless, the relapse rates remained exceptionally low throughout the follow-up period: 1.9 % in 50 months in the first (including only low-/intermediate-risk patients)50 and 1 % in 4 years in the second (including patients of all risk levels).34 Additionally, the S0521 cooperative study aimed to evaluate the role of maintenance in newly diagnosed low- and intermediate-risk patients who received intensive post-induction therapy containing ATO. Unfortunately, the study was prematurely closed due to low enrollment and after a median follow-up of 36.1 months, no relapses were observed in any of the groups. OS for the entire cohort was 93 %.51
In summary, the current recommendations regarding maintenance depend on risk stratification and the use of chemotherapy or ATO in addition to ATRA. For patients classified as intermediate-/low-risk treated with a protocol using ATRA plus chemotherapy, the role of maintenance is still controversial due to low relapse risk, especially for those achieving complete molecular remission at the end of consolidation. However, for patients classified as high-risk, maintenance seems to still play an important role in reducing relapse rates. Furthermore, maintenance is not recommended for intermediate-/low-risk patients treated with a protocol using ATRA plus ATO as relapse has become a rare event in randomized studies with long-term follow-up. Currently, the role of maintenance in high-risk patients treated with ATRA plus ATO is not clear, and studies analyzing this setting are ongoing.
Treatment complicationsDifferentiation syndromeDS is a common and life-threatening condition that can occur as a complication of treatment with ATRA or ATO. Both therapies induce the differentiation of leukemic blasts into mature cells and DS is an undesirable effect of these therapies. With an incidence of 2.5–37 % in patients treated with ATRA or ATO, it was first described in 1992.46,52–57
DS is characterized by a systemic inflammatory response and capillary leak syndrome. Differentiation of APL blasts induces the production of chemokines in the lung and ATRA stimulates leukemic cells to migrate towards alveolar epithelial tissues.58 Other inflammatory cells are attracted to this process, exacerbating it. Furthermore, by inducing the expression of beta integrins ATRA facilitates blast adhesion to the endothelium and subendothelium. Necropsy findings show diffuse neutrophilic infiltration and alveolar hemorrhage in the lungs, in addition to leukemia infiltration in several organs.59–60
Clinically, DS presents as dyspnea, pulmonary infiltrate on X-ray, fever, pleural or pericardial effusion, acute renal failure, hypotension, weight gain and edema.53 Infectious and cardiac etiologies must be ruled out, but if suspected, treatment must be initiated promptly. More rare manifestations include myocarditis, pericarditis, chest pain, pancreatitis, myalgia, and ocular manifestations with decreased visual acuity, retinal hemorrhage, and macular edema.
DS generally begins 7–12 days after starting ATRA treatment but can occur in around 40 days. About 50 % of cases occur in the first week of treatment and 50 % from the third week. Laboratory changes include leukocytosis, increased creatinine, and changes in troponin and pro-brain natriuretic peptide. Pulmonary infiltrate can be seen on chest X-ray in 80 % of severe cases. Pulmonary infiltrate, pleural effusion and ground-glass images or small pulmonary nodules can be seen by chest computed tomography. Some studies have shown that a WBC count >5–10 × 103/µL, elevated creatinine, and presence of coagulopathy are prognostic factors for the development of DS. In a recent publication, DS was involved in 12 early deaths (5.6 %) and thus was the second leading cause. Of these deaths, four resulted from DS alone, and eight were due to a combination of causes. The median time to DS-related early deaths was 10 days (range: 2–18 days). DS-associated deaths mainly occurred during the first weeks (10/12–83.3 % of patients).61 An incidence of DS was 5 % (18/180 patients) was reported in the Brazilian IC-APL study.30
Steroids are used to prevent DS, but there is no prospective evaluation of this benefit. Some protocols indicate prophylaxis if WBC count >5 × 103/µL or elevated creatinine (>1.4 mg/dL). A study from the Australian group used 1 mg/kg prednisone daily as prophylaxis for all patients treated with ATO and there was only one case of DS.43
Cytoreductive agents (hydroxyurea, chemotherapy or GO) can be used to control leukocytosis. The use of leukapheresis is not indicated due to the risk of hemorrhage.
Treatment must be started immediately if DS is suspected. In general, dexamethasone 10 mg every 12 h is used, in conjunction with treatment for infectious conditions and heart failure, when suspected. If there is no improvement after 24 h, the dose of dexamethasone can be increased to 10 mg every 6 h. ATRA and ATO should only be temporarily interrupted if the DS is severe, with severe respiratory failure, renal failure or when the patient requires admission to an intensive unit. After the condition has resolved, treatment with ATRA or ATO can be continued normally, within the protocol used to treat APL. Dexamethasone should be maintained until complete disappearance of signs and symptoms. Other support measures must be associated to treat signs or symptoms of fluid overload, dialysis, including mechanical ventilation, depending on the severity of each case.62
Pseudotumor cerebriPseudotumor cerebri (PTC) is a rare complication of ATRA in APL treatment, occurring more frequently in children.63–71 In a large series of 1034 APL patients enrolled in the PETHEMA Group trials, (APL96, APL99 and APL2005), the incidence of PTC was 3 % (30 cases during induction and 2 cases during consolidation). There was a higher rate of PTC in patients under 18 years old (13 %) and from 18 to 25 years, while in over 50-year-old patients it was 0.3 %. The rate of recurrence was 28 % after reinitiating ATRA.72 In 240 APL patients from the Intergroup Protocol 0129 (I0129), probable PTC occurred in 1.7 % of patients (ages: 4–24 years old) who received ATRA 45 mg/m2/day during induction or maintenance. The symptoms began 2–19 days after starting ATRA.73 There are also reports of PTC in patients treated with the ATRA plus ATO combined regimen74 and rare reports of PTC secondary to ATO.75
The following signs and symptoms suggest a diagnosis of PTC: headache, blurred vision, diplopia, and nausea and vomiting with bilateral papilledema characterized by increased intracranial pressure and the presence of papilledema, normal or diminished ventricles and normal cerebrospinal fluid (CSF). Diagnosis is made by the presence of papilledema, normal neurologic exam except for cranial nerve abnormalities, normal neuroimaging without evidence of hydrocephalus, mass, or structural lesion, and no abnormal meningeal enhancement on magnetic resonance images or by contrast-enhanced computed tomography, normal CSF composition, and elevated lumbar puncture opening pressure (≥250 mmHg or ≥280 mmHg for non-obese children). The diagnosis may also be made in the absence of papilledema if all of the above criteria are satisfied and the patient has unilateral or bilateral abducens nerve palsy. If there is neither papilledema nor a sixth cranial nerve palsy, the diagnosis can be suggested, but not confirmed with additional neuroimaging characteristics. Headache, although a common symptom, is not one of the criteria.73,76 In all cases there was an improvement of symptoms in 2–28 days after the suspension of ATRA, which was often reinitiated at a lower dose. Other therapies that have been utilized include corticosteroids, acetazolamide, mannitol, glycerin, topiramate, and therapeutic lumbar puncture.71,73,77 There is a report of the use of mannitol and acetazolamide together with the suspension of ATRA to decrease the CSF pressure thereby improving the headaches, diplopia, nausea and vomiting in two days and improvement of papilledema in seven days.78 Topiramate, an anticonvulsant and carbonic anhydrase inhibitor, may also be used in cases not responsive to acetazolamide to improve symptoms.74
The temporary suspension of ATRA/ATO is recommended when there is diagnostic suspicion of PTC in patients with APL. Symptomatic drugs may be used to abbreviate symptoms. ATRA/ATO can be reintroduced after improvement of the symptoms.
Response assessment and monitoringMorphologic assessment post-inductionIn the era of ATRA based treatment regimens, induction failures (failure to achieve morphological response) usually account for less than 1 % of cases.40,79–81 Morphological remission (less than 5 % of marrow blasts) should be confirmed at blood count recovery (platelet >100×103/µL and neutrophils > 1.5 × 103/µL)7 or 28 days after starting induction if no sign of count recovery.
Measurable residual diseaseMeasurable residual disease (MRD) negativity after consolidation therapy is an essential goal of treatment. Due to kinetics and terminal differentiation, post-induction PML::RARA positivity is common and should not change clinical decisions. In a phase 3 study that compared ATRA plus ATO and ATRA plus Idarubicin, 27 % of patients (20 out of 74) in the ATRA plus IDA group remained MRD positive while 44 % (36 out of 82 maintained RT-qPCR positivity 60 days after induction.34 The adequate timepoint for assessing MRD using RT-PCR or RT-qPCR is at marrow recovery at the end of consolidation. Persistence of MRD positivity after consolidation or conversion from MRD negative to positive as identified by RT-qPCR is associated with morphologic relapse and is an important tool to predict relapse free survival while allowing for preemptive salvage treatment.82,83
RT-qPCR is the preferable testing method because of higher sensitivity; it is also less prone to contamination issues and easier to standardize.84 Once molecular response is confirmed, the European LeukemiaNet guidelines for the management of APL state that monitoring for relapse is cost effective principally for high-risk patients (WBC count >10 × 103/µL).82 Accordingly, the European LeukemiaNet MRD consensus document suggests monitoring every three months for 2–3 years after the end of consolidation only for high-risk APL. A minimum detection threshold of one mutated cell in 1000 cells (0.1 %) is recommended.85
Preferred sampleBone marrow remains the preferred sample for MRD testing, because of higher sensitivity and earlier positivity when compared to peripheral blood.82 However, peripheral blood is a reasonable alternative if used in shorter intervals (every 4–6 weeks). In the case of MRD conversion from negative to positive or when a 1log10 elevation occurs as determined by RT-qPCR in patients that were previously stable, the result must be confirmed using a bone marrow sample at least two weeks after.7,85,86
Special situationsAcute promyelocytic leukemia in childrenAPL occurs in 0.06 cases per 100,000 per year in the pediatric population. This represents 5–10 % of cases of AML in this population. It shows gender equivalence. Similar to adults, it occurs more frequently in certain populations such as those in Latin America, China, Spain, and Italy. Under 5-year-old children have worse outcomes compared to those over 5 years old.87,88
Treatment regimens are based on those performed in adults with certain adaptations in the doses of ATRA due to the increased risk of neurological complications, such as pseudotumors, which occur in 15 % of cases compared to 3 % in adults. Therefore, in all protocols using ATRA in combination with chemotherapy, the dose is reduced to 25 mg/m2/day. Several multicenter studies, notably the Italian group (GIMEMA) and the Spanish group (PETHEMA), using ATRA in combination with idarubicin for induction followed by 3 cycles of consolidation associated with anthracyclines and maintenance for two years with mercaptopurine, methotrexate, and ATRA, showed excellent results. In the GIMEMA study, 96 % of patients achieved CR with PML::RARA negativity at the end of consolidation. OS was 89 %, and DFS was 75 %. Of note, WBC count >10 × 103/µL significantly reduced DFS.7,89
In this context, the PETHEMA group suggested that their regimen used until 1996, with an induction scheme of ATRA 25 mg/m2 divided in two doses until CR or 90 days, and idarubicin 12 mg/m2/day on Days 2, 4, 6, and 8 followed by consolidation (cycle 1: idarubicin 5 mg/m2/day on Days 1 to 4; cycle 2: mitoxantrone 10 mg/m2/day on Days 1 to 5; cycle 3: idarubicin 12 mg/m2/day on Day 1) was reinforced for higher-risk cases after 1999 during consolidation by the addition of ATRA 25 mg/m2 for 15 days and an increase in the idarubicin dose to 7 mg/m2/day on Days 1 to 4 in the first cycle and two consecutive days in the third cycle. After completing consolidation with PML::RARA negativity by RT-PCR, maintenance with mercaptopurine 50 mg/m2/day, methotrexate 15 mg/m2/week, and ATRA 25 mg/m2/day for 15 days every three months was carried out for two years, adapting doses to hematologic levels and changes in transaminases. The results showed an OS of 91 % ± 8 % and DFS of 89 % ± 12 %.90
An important concern in children is to reduce cardiotoxicity, which is linked to secondary neoplasms, because of their long life expectancy. A study from the International Consortium for Childhood APL evaluated a reduction in the dose of anthracyclines from 650 mg/m2 to 355 mg/m2 for standard risk and to 405 mg/m2 for high risk without a significant change in outcomes.87
As in adults, several studies have demonstrated the safety and efficacy of the combination of ATO and ATRA in children. The main difference from adults is the reduction of the ATRA dose to 25 mg/m2 following the same scheme adopted by the APL0406 study of adults. In high-risk patients, the use of low doses of anthracyclines or GO plays a crucial role. Additionally, a Chinese study has shown excellent results with an oral formulation of ATO.34,91,92
Acute promyelocytic leukemia in older patientsThe management of APL is particularly challenging in elderly patients. General supportive measures should be given paying special attention due to the high risk of early mortality. According to Swedish registry data, early mortality rates reach up to 50 % among over 60-year-old patients compared to 15 % in younger patients.93
Within this context, the ATRA plus ATO combination is assumed to be even more beneficial for older patients due to its lower hematological toxicity and infection rates. This comes from limited, retrospective data, since over 70-year-old patients are infrequently included in clinical studies.94 According to the AML17, the arsenic dosage can be safely adjusted to twice weekly to prevent toxicity caused by this drug.34
Due to their underrepresentation in clinical studies, there is greater controversy regarding the optimal strategy for high-risk APL. In such cases, geriatric assessment could prove crucial and should replace chronological age as a decision-making factor. The assessment of geriatric domains, such as physical capacity, functionality, and comorbidities, will identify the more vulnerable patients who may benefit from exclusive treatment with ATRA plus ATO or from a combination with reduced doses of anthracyclines. The use of GO may be particularly appropriate for patients with a high risk for cardiac toxicity. Conversely, elderly patients without abnormalities in geriatric domains and compensated comorbidities should be considered for standard high-risk APL treatment, optimizing outcomes related to the control of the leukemia.95
Acute promyelocytic leukemia during pregnancyThe diagnosis of acute leukemia during pregnancy is a rare and dramatic condition, with 1:100,000 pregnancies described.96 APL during pregnancy is even more challenging due to its clinical characteristics that include the potential coagulopathy that may pose serious risks for both mother and fetus. The incidence is not well established and only based on rare case reports.97,98 A recent systematic review included 53 reports of individual cases, among which 92 patients were eligible for induction therapy, either with ATRA (32 %), ATRA and anthracycline (43 %), or anthracycline only;98 the CR rate was 89 %.98,99
Besides the increased risk of maternal and fetal death, intrauterine growth restriction, and malformations are additional risks when APL and pregnancy coexist so that the discussion needs to be discussed by the patient, family, and the medical multidisciplinary team.6,7,97,98 Medical termination of pregnancy is usually contraindicated upon diagnosis due to pancytopenia and coagulopathy, which are not controlled at that moment.6,7
If APL is diagnosed in the first trimester, therapeutic options are quite limited, and the risk of miscarriage is higher compared to the second or third trimesters (88 % versus 30 %).98 Although specific information regarding the teratogenicity of ATRA is not available, this effect is well demonstrated with other retinoids6 and, therefore, it should be avoided in the first trimester. Although anthracyclines may also increase the risk of miscarriage, fetal malformations, and low birth weight, chemotherapy is needed with the recommended anthracycline being daunorubicin, due to its well-known efficacy in APL, and the limited evidence against the use of idarubicin that has a greater ability to cross the placental barrier.6,100 Considering the benefits of the ATRA association with chemotherapy, the appropriate timing for introducing ATRA should be discussed individually.6
In the second and third trimesters, ATRA should be initiated immediately, as has been recommended by the European LeukemiaNet since 2009.6 It can be used with relative safety in the second and third trimesters of pregnancy with no cases of fetal malformations, although delayed fetal growth and fetal arrhythmia have been reported.101
ATO is genotoxic, teratogenic, and carcinogenic, as demonstrated by studies in animal models and environmental risk studies.99,102,103
Since it crosses the placental barrier, it is recommended to avoid its use throughout pregnancy.6 However, a report has described the use of ATRA and ATO during pregnancy with a favorable outcome for both mother and fetus, suggesting that this is a question of debate considering that this combination is currently the best therapeutic approach for APL.104
Another challenge is the choice of delivery type, which must be based on each patient's conditions. Vaginal delivery should be pursued whenever possible, as the risk of bleeding is higher in cesarean section.104
In summary, APL during pregnancy carries a high probability of miscarriage, prematurity, low birth weight, and respiratory distress syndrome at birth.98 Fetal viability is strongly influenced by gestational age, with a mortality rate of 87 % in the first trimester, 33 % in the second, and 7 % in the third.97,98 Pregnant women with adequate prenatal care can be diagnosed early and may have a better prognosis.
In conclusion, despite the demanding management, a multidisciplinary approach and careful monitoring of coagulopathy may offer a chance for CR and cure, regardless of gestational age at diagnosis.
Management of relapseAlthough APL is a curable disease in more than 90 % of the cases, approximately 10–25 % of adult patients may relapse after achieving CR.105
Molecular relapse is defined as two successive RT-PCR assays, with stable or rising PML::RARA transcript levels detected in independent samples analyzed, ideally in two different laboratories. It normally precedes hematological relapse and carries better outcomes when treatment is started early (preemptive therapy). Hematological relapse occurs when more than 20 % of blasts/atypical promyelocytes are detected in a single BM sample or more than 5 % of blasts/atypical promyelocytes are present in two BM samples two weeks apart. Genetic confirmation should be performed with any of the methods previously described (RT-PCR, FISH, or conventional karyotype). Extramedullary relapse is a rare phenomenon (3–5 % among all relapses) and can affect any organ.106 Special attention must be given to the CNS, the most affected site. The incidence of extramedullary manifestations has been rising in the era of ATRA therapy. Some authors postulate that this can be attributed to the effect of ATRA on adhesion molecules which may result in increased infiltrative capacity of APL blasts at the sanctuary sites.107 Documentation in skin, CNS (either imaging or spinal fluid), or other sites is required as well as genetic confirmation by any of the conventional methods.
Selecting salvage therapy for molecular or hematologic relapse should consider the initial first-line treatment administered. Therefore, individuals experiencing relapse after ATRA plus chemotherapy should undergo treatment using an ATRA plus ATO-based approach until achieving MRD negativity. Conversely, for those who relapse follows ATRA plus ATO treatment, an ATRA plus chemotherapy approach is the appropriate choice. An exception to switching to an alternative initial treatment for relapsed patients can be considered for those encountering a late relapse, defined as a relapse occurring after more than two years in CR.
The main objective of salvage therapy is to achieve molecular remission as a bridge to hematopoietic stem cell transplantation (HSCT). Autologous HSCT should be considered as the first choice for eligible patients achieving second molecular remission (undetectable MRD in the BM).7,108,109 However, a recent report questions the role of transplantation, at least in patients achieving molecular remission with ATO and ATRA who do not have CNS disease at relapse and who have received a full course of consolidation with ATO.33
Chakrabarty et al.110 reported on 294 patients who received either allogeneic transplant (n = 232) or autologous transplant (n = 62) between 1995 and 2006. The 5-year DFS in the autologous transplant recipients was 63 % (range: 49–75 %) versus 50 % (range: 44–57 %) in patients receiving allogeneic transplant (p-value = 0.1), and OS was 75 % (range: 63–85 %) versus 50 % (range: 48–61 % - p-value = 0.002). Treatment-related mortality in patients receiving allogeneic transplant (30 %) was higher compared to autologous transplant (2 %). Therefore, the choice for autologous HSCT in MRD-negative patients has been based on studies showing less toxicity and similar survival as compared to allogeneic transplantation. In contrast, patients failing to achieve molecular remission are candidates for allogeneic HSCT with a matched sibling or alternative donor (haploidentical, unrelated donor or cord blood) (Figure 1).

Diagnosis and follow-up workup in acute promyelocytic leukemia. †Can be used for fast diagnosis. * Monthly PML::RARA from peripheral blood is an alternative to bone marrow's evaluation. ‡If RT-PCR positive or 1 log elevation in RT-qPCR, test should be repeated in a new bone marrow sample (at least 2 weeks apart).
Patients unsuitable for HSCT and those with a very prolonged CR can be managed with repeated cycles of ATO (e.g. 6 cycles) with or without ATRA and with or without chemotherapy, considering previous treatments and clinical conditions. For patients with CNS relapse, induction treatment consists of weekly triple intrathecal therapy (ITT) with methotrexate, hydrocortisone, and cytarabine until complete clearance of blasts in the cerebrospinal fluid, followed by 6–10 more spaced out ITT cycles as consolidation.7
Conclusions - current outcomes and perspectivesAPL is a very common subtype of AML in Latin American countries, unique for its maturation arrest in promyelocytes and close association with the characteristic molecular alteration, t(15;17)(q24.1;q21.2), along with a pronounced tendency towards coagulopathy, bleeding, and early death. Knowledge of its pathophysiology over the years has resulted in revolutionary differentiating therapies (ATRA since the 1990s and ATO since 2013). With modern combined therapy, APL has become a curable acute leukemia even with a chemotherapy-free regimen. Considering such success, paradoxical to the risk of early death, the recognition of APL is imperative.
In this context, a global initiative by the American Society of Hematology with the leadership of Brazilian experts culminated in an International Consortium of APL (IC-APL), established in 2005, initially including four Latin American countries (Brazil, Chile, Mexico, and Uruguay). The establishment of IC-APL resulted in a nearly 50 % decrease in early mortality and an improvement in OS of almost 30 % compared to historical controls, leading to survival rates similar to those reported in developed countries.111 In the latest update of this study, including 806 patients from Latin American countries (Brazil, Chile, Mexico, Uruguay, Peru, and Paraguay), already submitted for publication as of writing this consensus, the improvement in outcomes has been sustained over the past 15 years, with early mortality still the main cause of therapeutic failure.
More recently, a less toxic therapy with two targeted drugs, ATRA and ATO, has been encouraged and may be considered one of the greatest advances in AML treatment ever seen. Current international recommendations include this chemotherapy-free therapy for low-risk patients and suggest combining it with anthracyclines for high-risk patients as the first choice in APL treatment (Figure 2). When ATO is not available, the combination of ATRA and anthracyclines is feasible. Besides avoiding chemotherapy, the ATRA and ATO combination has shortened the duration of APL treatment since no maintenance with low-dose chemotherapy is needed. Future challenges still remain, especially in low and middle-income countries, including rapid diagnosis to treat patients promptly and reduce early mortality, implementing low-toxicity treatment in healthcare services, and preventing relapse in the high-risk group.
 management according to the Brazilian Hematology Committee recommendations. WBC: white blood cell; ATRA: all-trans-retinoic acid; ATO: arsenic trioxide; DS: differentiation syndrome; 6-MP: 6-mercaptopurine; MTX: methotrexate; HSCT: hematopoietic stem cell transplantation.")
Overview of acute promyelocytic leukemia (APL) management according to the Brazilian Hematology Committee recommendations. WBC: white blood cell; ATRA: all-trans-retinoic acid; ATO: arsenic trioxide; DS: differentiation syndrome; 6-MP: 6-mercaptopurine; MTX: methotrexate; HSCT: hematopoietic stem cell transplantation.



