





The guidelines project is a joint initiative of the Associação Médica Brasileira and the Conselho Federal de Medicina, aiming to bring information in medicine to standardize management and assist in decision-making during treatment. The recommendations of this article were elaborated by the Associação Brasileira de Hematologia, Hemoterapia e Terapia Celular (ABHH). The attending physician should evaluate all possible medical approaches, considering the patient characteristics and clinical status.
This article presents the guidelines on sickle cell disease: secondary stroke prevention in children and adolescents.
Description of the method used to collect evidenceThese guidelines result from a systematic evidence-based review centered on the Evidence-Based Medicine movement, in which clinical experience is integrated with the ability to critically analyze and apply scientific information rationally, thereby improving the quality of medical care.
The questions were structured using the Patient/Problem, Intervention, Comparison and Outcome (PICO) system, allowing the generation of evidence search strategies in Medline (via PubMed) and using a manual search. The data recovered were critically analyzed using discriminatory instruments (scores) according to the type of evidence. After identifying studies that potentially substantiated the recommendation, the level of evidence and degree of recommendation were calculated using the Oxford Classification.1
BackgroundThe genetic hemoglobin (Hb) mutation caused by the replacement of glutamic acid by valine in position 6 of the beta-globin chain (β6 Glu → Val) produces the variant hemoglobin S (HbS → α2β2s). Sickle cell disease (SCD) is a group of hereditary autosomal recessive hemoglobinopathies. Sickle cell anemia (SCA, homozygous HbS or HbSS) is the most common and severe manifestation of SCD and double heterozygosis occurs as HbS/β0-thalassemia (HbSβ0, severity as HbSS), HbS/β+-thalassemia (HbSβ+), HbSC disease (HbSC), HbSD disease (HbSD) and other less frequent associations.2(D)
The SCD physiopathology is based on anemia and vaso-occlusive events caused by HbS polymerization, a process dependent on hypoxia that leads to inflammation, excessive adhesion of erythrocytes to the activated endothelium, ischemia-reperfusion injury, a hypercoagulable state, and dysregulation of the vascular tone.2(D)
Stroke is a significant cause of morbidity and mortality in individuals with SCD, leading to severe motor and neurocognitive sequelae.3(B),4(D) Stroke physiopathology can be explained by vasculopathy and occlusion caused by the sickle erythrocytes, with stenosis of cerebral vessels of the circle of Willis (middle cerebral artery, anterior cerebral artery, anterior communicating artery, internal carotid artery, posterior cerebral artery and posterior communicating artery).5(D) Patients with SCA develop vasculopathy and occlusion at specific sites, such as the distal internal carotid artery and the proximal segments of the middle cerebral artery and anterior cerebral artery.6,7(A)
An acute episode of ischemic stroke in patients with SCD is treated with blood transfusion to decrease HbS.4(D) Without secondary stroke prevention, more than half of patients who experienced the first episode will have a recurrence.3(B) Observational studies have established that chronic packed red blood cell (pRBC) transfusions decrease the risk of a new stroke.8–11(B)
Clinical questions1. Is there evidence that red cell exchange transfusion is the treatment of choice for acute ischemic stroke in patients with sickle cell disease?
Acute ischemic stroke is a severe complication in patients with SCA, occurring in 10 to 11% of patients under 20 years old.6,12(A) The predictive risk of an initial stroke during the first two decades is 0.761 episodes per 100 patient-years.3(B)
Hulbert et al., in a retrospective observational study on 124 children with SCA and data on the duration of stroke symptoms, assessed which would be the most effective transfusion treatment for an acute ischemic stroke and to decrease the risk of a subsequent stroke: simple blood transfusion or exchange transfusion (manual or erythrocytoapheresis). The mean age at a first stroke was 6.3 years (range 1.4–14 years) and the mean follow-up was 10.1 years (range 5–24 years). Patients should have been undergoing chronic pRBC transfusions for at least five years after the first stroke. Red cell exchange transfusion was the most common initial treatment for the first stroke, regardless of the duration of symptoms. The initial transfusion treatment was available for 52/80 (65%) patients who came to medical care within 24 h of the stroke symptoms. In patients initially treated with a simple red blood cell transfusion, recurrent stroke occurred in 57% (8/14), compared to 21% (8/38) in those treated with red cell exchange transfusion. Children with SCA and first acute stroke receiving simple blood transfusion had a 5-fold greater relative risk of the second stroke in comparison to exchange transfusion (RR = 5, 95% confidence interval (CI) 1.3–18.6, p = 0.02). Patients without medical history (fever, acute anemia, acute chest syndrome or high blood pressure) prior to the acute stroke who received simple red blood cell transfusion had an 8-times greater risk of a second stroke, compared to patients treated with exchange transfusion (RR 8.0; 95%CI 1.7–3.8; 8/11 patients versus 7/28 patients, respectively). Study limitations are the retrospective design, lack of the possibility of establishing a cause-and-effect relation between the type of transfusion and the subsequent stroke and the small number of second stroke, not allowing multivariate analysis.13(B)
For ethical reasons, there is no prospective controlled clinical trial comparing exchange transfusion and simple transfusion as initial acute ischemic stroke treatment in children with SCA. Treatment guidelines are written based on the published literature.4(D)
Simple transfusion or exchange transfusion (manual or erythrocytoapheresis) reduce acute sickling and improve cerebral blood flow.14(D) The transfusion type depends on individual patient factors and local transfusion resources (strong recommendation based on high certainty of evidence on effects). The primary objective of the exchange transfusion is to reduce HbS < 30%, with a target Hb = 10 g/dL, without increasing blood viscosity. If the exchange transfusion is not available within 2 h and the Hb ≤ 8.5 g/dL, a single transfusion can be prescribed to avoid delays in treatment until the exchange transfusion is available (conditional recommendation based on low certainty in the evidence on effects). When a simple transfusion is prescribed, it is important to pay attention to the final Hb (maximum Hb = 10 g/dL).4(D)
In children with SCA and acute ischemic stroke, while awaiting the pRBC for simple transfusion or exchange transfusion, the recommendations are:515(D)
- •
Immediate intravenous hydration: may contribute to the improvement of stroke signs and symptoms.
- •
Puncture a good vein access.
- •
Phenotyped and filtered PRBC, including routine procedures (ABO and Rh typing and irregular antibody screening).
- •
Support measures and maintain oxygen saturation > 95%.
- •
Magnetic resonance imaging (MRI) or cranial tomography (when MRI is not available) in the first hours and after the patient stabilization. Consider transient ischemic attack, convulsive disorders or hemiplegic migraine, when the image is normal.
- •
Multidisciplinary approach to acute ischemic stroke with a pediatric hematologist, a neurologist and transfusion medicine physicians.
Recommendations
There is a lack of prospective, randomized, and controlled studies to manage acute ischemic stroke in children with sickle cell anemia (SCA). The only available study (retrospective) concluded, albeit with limitations, that patients who present signs and symptoms of an acute episode of ischemic stroke should undergo exchange transfusion (manual or automated erythrocytoapheresis) in the acute episode. The aim is to achieve HbS < 30% and hemoglobin not exceeding 10 g/dL. In this situation, exchange transfusion may prevent the recurrent stroke in children with SCA more than a single transfusion.
In the absence of manual or automated erythrocytoapheresis, or when Hb ≤ 8.5 g/dL, an option with a lower level of evidence would be simple transfusion.
2. Is there evidence that chronic packed red blood cell transfusions can prevent a new stroke episode (secondary prevention) in patients with sickle cell disease?
Powars et al. demonstrated in a single-center, retrospective, cohort study the high stroke recurrence rate in patients with SCD (33 HbSS and 2 HbSC) when no treatment was administered, both in the 3 and 9 years after the first stroke (80% and 66%, respectively).3(B) Retrospective single-center studies, with few pediatric patients with SCD who had a previous stroke, observed a decrease in stroke recurrence when on regular exchange transfusion to maintain HbS < 30% and Hb around 10 g/dL.8,16,17(C)
Russell et al. studied the effect of transfusion therapy on arteriographic abnormalities and stroke recurrence in 30 pediatric patients with SCA. The objectives were to analyze the cause of acute neurological deficits and assess the effects of pRBC transfusions administered for at least one year after the acute stroke episode. Prolonged transfusion therapy to maintain HbS < 30% nearly stopped the progression of stenosis and markedly decreased the irregularity of the luminal surfaces, decreasing to 10% the stroke recurrence rate (p < 0.001), despite persistent arterial abnormalities.9(B)
In a multicenter, retrospective cohort study published by Pegelow et al., 60 patients with SCD (57 HbSS, 2 HbSβ+ and 1 HbSβ0), aged 20 months to 24 years at the time of the first ischemic stroke and receiving long-term transfusion therapy to maintain HbS < 30%, were followed for a cumulative observation time of 191.7 patient-years. The mean follow-up was 38 months, with a maximum follow-up time of 71 months. Patients who received chronic transfusion therapy after a stroke were less likely to have a recurrence (p < 0.001). The stroke recurrence was 4.2 per 100 patient-years. There was a significant reduction in the stroke incidence, compared to historical controls without regular pRBC transfusions.4(D),10(B)
In 10 children with SCA and previous stroke, when pRBC transfusions to maintain HbS < 20% were stopped after 1 or 2 years of treatment, the recurrence rate was similar to that found in non-transfused patients with previous stroke. The second stroke occurred 5 weeks to 11 months after stopping transfusions (median 3 months) in 7/10 patients.18(C) Similar results were found in another 10 children with SCA and previous stroke (median 9½ years, range 5–12 years) after stopping pRBC transfusions (HbS < 30%): high recurrence rate within 12 months. In 5/10 patients, an ischemic event occurred, 4 with HbS 61 to 90%.19(C)
The current practice at different hematology centers is to continue transfusion therapy indefinitely. Therefore, complications related to alloimmunization and iron overload become real.20(B),21(A)
To decrease the transfusion iron burden, Cohen et al. proposed a chronic transfusion regimen to maintain HbS < 50%. The study included 15 patients (10–21 years, mean 15 years) with SCA and previous stroke being treated with simple chronic transfusions to maintain HbS < 30% for at least 4 years and without recurrence. After a median follow-up of 84 months (14–130 months), no patient with pre-transfusion HbS < 50% had recurrent cerebral infarction in 1,023 patient-months. The single transfusion decreased transfusion requirements 17% to 48% (mean 31%, p < 0.001) and exchange transfusion (manual or automated) 33% to 99% (mean 67%, p < 0.001). Considering the importance of iron overload control with iron chelation therapy, by reducing the transfusional requirements, the iron burden also decreases.22(C)
Miller et al. proposed a less intensive transfusion regimen (HbS < 60%) to prevent recurrent stroke in SCA patients who were on regular transfusions to maintain HbS < 30% for a mean of 9 years. In 13 patients, after at least one year (12–27 months, mean 15.5 months) of follow-up, there was no recurrent neurological event and two patients died from complications of transfusional hemochromatosis. The reduction in transfusion requirements during the first year was 31.4%.23(C)
These results suggest that HbS < 50 to 60% could be a target, but the sample size is too small to conclude that this level is as effective as HbS < 30% to prevent recurrent stroke. Thus, the recommendation for pre-transfusional Hb S < 30% should be maintained.4(D)
The partial manual exchange (Box 1) reduced serum ferritin during pRBC transfusions in 17 children with SCA (median 8.1 years, range 0.7–16.8). They received 487 transfusions (median 29, range 18–49) before the initial liver iron concentration (LIC) assessment. Serum ferritin decreased -8.8 ng/mL for each mg/kg of iron phlebotomized (p = 0.02). Although a reduction in LIC from pre-transfusional phlebotomy could not be established (p = 0.4), partial manual exchanges appear to be an effective strategy for decreasing the iron burden.24(B)
Box 1. Partial manual exchange, considering a red cell unit hematocrit of 65%.
Hb: hemoglobin. Adapted from Savage WJ, 2013.24(B)
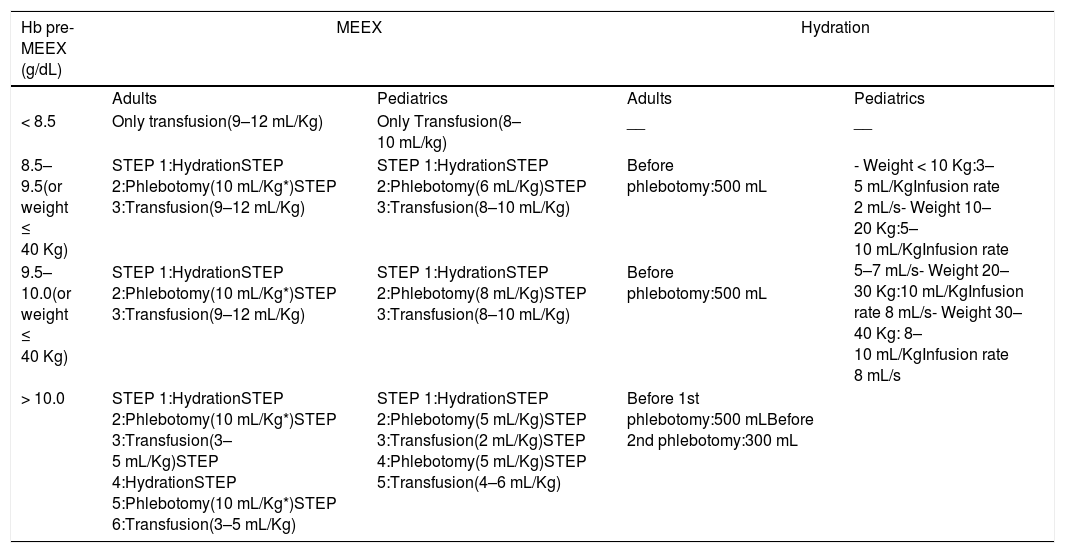
A prospective multicenter study validated a manual exchange transfusion protocol (Table 1), in which volume and final Hb are estimated. Thus, it is possible to predict the percentage of HbS after the procedure. The protocol was evaluated in 261 manual exchange transfusions in SCD (HbSS and HbSβ) patients. It showed a correlation between the estimated and measured values of HbS and Hb (Pearson correlation coefficient Rp 0.95 and 0.65, respectively, p < 0.001). The Rp was used to evaluate the correlation between variables. This procedure was shown to be safe (no adverse events) and effective with a single venous access.25B)
Procedural scheme for Manual Erythroexchange (MEEX).
| Hb pre-MEEX (g/dL) | MEEX | Hydration | ||
|---|---|---|---|---|
| Adults | Pediatrics | Adults | Pediatrics | |
| < 8.5 | Only transfusion(9–12 mL/Kg) | Only Transfusion(8–10 mL/kg) | __ | __ |
| 8.5–9.5(or weight ≤ 40 Kg) | STEP 1:HydrationSTEP 2:Phlebotomy(10 mL/Kg*)STEP 3:Transfusion(9–12 mL/Kg) | STEP 1:HydrationSTEP 2:Phlebotomy(6 mL/Kg)STEP 3:Transfusion(8–10 mL/Kg) | Before phlebotomy:500 mL | - Weight < 10 Kg:3–5 mL/KgInfusion rate 2 mL/s- Weight 10–20 Kg:5–10 mL/KgInfusion rate 5–7 mL/s- Weight 20–30 Kg:10 mL/KgInfusion rate 8 mL/s- Weight 30–40 Kg: 8–10 mL/KgInfusion rate 8 mL/s |
| 9.5–10.0(or weight ≤ 40 Kg) | STEP 1:HydrationSTEP 2:Phlebotomy(10 mL/Kg*)STEP 3:Transfusion(9–12 mL/Kg) | STEP 1:HydrationSTEP 2:Phlebotomy(8 mL/Kg)STEP 3:Transfusion(8–10 mL/Kg) | Before phlebotomy:500 mL | |
| > 10.0 | STEP 1:HydrationSTEP 2:Phlebotomy(10 mL/Kg*)STEP 3:Transfusion(3–5 mL/Kg)STEP 4:HydrationSTEP 5:Phlebotomy(10 mL/Kg*)STEP 6:Transfusion(3–5 mL/Kg) | STEP 1:HydrationSTEP 2:Phlebotomy(5 mL/Kg)STEP 3:Transfusion(2 mL/Kg)STEP 4:Phlebotomy(5 mL/Kg)STEP 5:Transfusion(4–6 mL/Kg) | Before 1st phlebotomy:500 mLBefore 2nd phlebotomy:300 mL | |
Maximum limit of single phlebotomy 700 mL, for a maximum limit of total phlebotomy of 1400 mL. Transfusion volume is calculated according to the hematocrit of the red blood cell unit 60%. Adapted from Gianesin B, 2020.25(B).
HbS% and Hb concentration after manual erythroexchange can be calculated based on the volumes exchanged during the procedure (Figures 1 and 2).25(B)
")
Formula to calculate the HbS% after the manual erythroexchange. Adapted from Gianesin B, 2020.25(B)
")
Formula to calculate the Hb g/dL after the manual erythroexchange. Adapted from Gianesin B, 2020.25(B)
A multicenter, retrospective cohort study in 137 children (SCA, previous stroke, chronic transfusion therapy for at least 5 years, HbS 30–50%) found a stroke recurrence rate of 22.6% or 2.2 events per 100 patient-years. The mean age at the first stroke was 6.3 years (1.4–14.0 years) with a mean follow-up of 10.1 years (5–24 years).11(B) In a prospective, multicenter, single arm study in 40 children with SCA, stroke and regular blood cell transfusions with a mean pre-transfusion HbS = 29%, followed for a median of 5.4 years (222 patient-years), the incidence of stroke recurrence was 17.5%, or 3.1 events per 100 patient-years.26(B)
Recurrences could be related to the HbS level, the severity of the cerebrovascular disease and the medical or concurrent events. Patients with no risk factor associated with their initial stroke had a higher risk of recurrence, despite long-term transfusions (p = 0.038).11(B) Progressive vasculopathy, such as severe arterial stenosis or Moya-Moya syndrome, contributes to the silent infarction or stroke, despite HbS < 30%.26(B)
Recommendations
1. For children and adolescents with sickle cell disease (HbSS or HbSβ0) and previous ischemic stroke, chronic packed red cell transfusions should be performed for secondary stroke prevention to maintain the pre-transfusional HbS level < 30% and Hb level between 9 and 10 g/dL.
2. Despite 20% of patients possibly having a new stroke while on chronic red cell transfusion and HbS 30 to 50%, this treatment remains the only proven and effective strategy for secondary stroke prevention.
3. Manual or automated exchanges are effective to maintain HbS < 30% and to decrease the iron burden.
3. Is there evidence that hydroxyurea can prevent a new stroke episode (secondary prevention) in patients with sickle cell disease?
Considering that chronic pRBC transfusions are the standard of care for secondary stroke prevention in children and adolescents with SCD, studies to investigate hydroxyurea (HU) efficacy to prevent secondary stroke are scarce, being case reports or observational studies.27–28(C),29–32(B)
Fester et al. observed that of the 9 pediatric patients with SCA and stroke or transient ischemic attack receiving HU, after a mean follow-up of 3.5 years, none had a new neurological event.27(C) In another study, of the 5 children with SCA, a previous stroke and HU 30 to 40 mg/kg/day, none had a recurrence during the follow-up (42–112 months).28(C)
In a prospective study by Ware et al., transfusions were discontinued in 16 pediatric patients with SCD (15 HbSS, 1 HbS/OArab) and previous stroke who had at least one of the following: erythrocyte alloantibodies or autoantibodies, recurrent stroke even in transfusion therapy, iron overload, lack of adherence to treatment or allergy to deferoxamine. The HU was escalated to the maximum tolerated dose (MTD) (mean 24.9 ± 4.2 mg/kg/day), and phlebotomy was offered to those with iron overload. A new stroke occurred in 3/16 (19%) patients 3 to 4 months after starting the HU and before reaching the MTD.29(B)
In another prospective study, Ware et al. discontinued chronic transfusions in 35 children with SCD (33 HbSS, 1 HbSβ0 and 1 HbS/OArab) who had a previous stroke and at least one of the following: multiple red cell alloantibodies, red cell autoantibodies, recurrent stroke despite transfusions, systemic allergy to deferoxamine, severe iron overload due to low compliance to chelation therapy, low compliance to the transfusion regimen, parental request, or religious objection to blood transfusion. The HU was started for secondary stroke prevention and phlebotomy, for iron overload. One group (n = 20) received concurrent transfusions and HU for a mean time of 6 ± 3 months (range 3–15) until reaching the HU MTD. Another group (n = 15) stopped transfusions and started HU and after a follow-up of 42 ± 30 months (range 3–104 months) at a HU dose of 26.7 ± 4.8 mg/kg/day, the stroke recurrence rate was 7.4 events/100 patient-years, while for the children who received transfusion until reaching the HU MTD, the stroke recurrence rate was 3.6 events/100 patient-years. Therefore, achieving the HU MTD before stopping blood transfusions is very important.30(B) In a retrospective analysis of this study, after a median follow-up of 5.6 years and a total of 219 patient-years, 10/35 (29%) patients had a new stroke after switching transfusions to HU, suggesting that long-term treatment with HU reduced, but did not eliminate, the risk of stroke recurrence.33(B)
The phase III, randomized, multicenter, non-inferiority clinical trial Stroke With Transfusions Change to Hydroxyurea (SWiTCH) compared standard treatment (pRCB transfusions + iron chelation; n = 66) with an alternative treatment (HU MTD + phlebotomy; n = 67) in children (mean age 13 years) with SCA and a previous stroke. The mean dose of HU was 26.2 ± 4.9 mg/kg/day. The study was terminated early because of the evidence that HU was less effective than regular pRBC transfusions in secondary stroke prevention. Patients under standard treatment had no stroke, while 7 patients in the alternative treatment developed a new stroke episode. However, this difference was not significant (RRA = -0.044 with 95%CI: -0.044–0.017).34(B)
The cerebral metabolic rate of oxygen (CMRO2) measures the cerebral oxygen demand, corresponding to the product of arterial oxygen content (CaO2), cerebral blood flow (CBF) and oxygen extraction fraction (OEF). In SCA patients, the CaO2 is low, so as to maintain the CMRO2, both CBF and OEF are elevated. pRBC transfusions increase the Hb, consequently increasing the CaO2 and decreasing the CBF and OEF, relieving the CMRO2 and stroke risk. To understand the action of the HU in the stroke prevention, a prospective study with 84 SCA patients (8–23 years old) showed that in those without HU (n = 23), the OEF was higher (median 42.9% [interquartile range (IQR) 39.1–49, 1%]) than in the HU treatment group (n = 38) (median 40.7% [IQR 34.9–43.6%]). Transfusions (n = 23) had the lowest OEF values (median 35.3% [IQR 32.2% -38.9%], p < 0.001). In addition, the percentage of white matter with the highest risk of ischemia was lower in the cohort with HU than without HU (p = 0.025 and p = 0.034, respectively), but higher than in the cohort with transfusions (p = 0.018 and p = 0.029, respectively). The brain metabolic stress (CMRO2) is reduced in patients with SCA receiving HU, compared to patients without HU, but is still higher than in patients with pRBC transfusions. The HU may offer neuroprotection by attenuating cerebral metabolic stress in SCA patients, but not to the same degree as transfusions.35(A)
In Jamaica, Ali et al. started the HU to 10/43 (23.6%) SCA children as secondary stroke prevention and one episode of stroke was observed (incidence rate 2/100 person-years). In contrast, in the 33 children with previous stroke (31 HbSS, 1 HbSC and 1 HbSb0) and no HU nor transfusions, there were 20 recurrences (incidence rate 29/100 person-years), with a hazard ratio of 9.4 (95%CI: 1.3–70.6; p = 0.03).36(B)
Lagunju et al. (Nigeria), in a retrospective study with 31 children (SCA and previous stroke) started the HU 20 to 25 mg/kg/day in 13 patients and observational treatment in 18 patients. The second stroke incidence was 7/100 patient-years in the HU group and 28/100 patient-years in the non-HU group (p = 0.001, OR 3.808, 95%CI 1.556–9.317). Thus, in regions where access to chronic blood transfusions is not possible or feasible, the HU therapy should be considered for secondary stroke prevention.31(B) Abdullahi et al. (Nigeria) provided the HU at a fixed dose of 20 mg/kg/day to 27 children with SCA as secondary stroke prevention due to poor blood availability, low parental acceptance of blood transfusion or transfusion cost. The recurrence rate was 17.4 events per 100 patient-years (95%CI 7.1–36.3 events per 100 patient-years), with two deaths.32(B)
In a meta-analysis published in 2020, where only the SWiTCH trial had the necessary inclusion criteria to answer whether HU could replace monthly blood transfusions in secondary stroke prevention, the conclusion was that for children with SCA and a history of cerebrovascular abnormality, transfusions should be used together with iron chelation therapy.37(D) However, a Cochrane Database of Systematic Reviews meta-analysis concluded that only the SWiTCH trial is not enough to establish that the HU and phlebotomy increased the risk of stroke [RR 14.78 (95%CI 0.86 -253.66)] or mortality [Peto OR 0.98 (95%CI 0.06–15.92)], compared to pRBC transfusions and iron chelation.38(D)
The British Society for Haematology Guideline recommends that transfusion therapy remain the standard of care for secondary stroke prevention in children with SCA. However, the HU can be recommended as second-line therapy when transfusions are contraindicated or unavailable (level of evidence 1B).39(D)
The American Society of Hematology recommends for secondary stroke prevention in children with HbSS or HbSβ0 exchange blood transfusion to raise the Hb level above 9 g/dL and maintain the HbS level < 30% (strong recommendation based on the low certainty in the evidence on effects). Chronic pRBC transfusions should be continued into adulthood. However, in cases where transfusion is not possible, the HU can be offered, knowing that the HU is inferior to chronic transfusions, but superior to no therapy in secondary stroke prevention.4(D)
Recommendations
1. Stroke is an important and frequent complication in children and adolescents with sickle cell anemia and the high recurrence rate must be prevented. There is no evidence to support the use of hydroxyurea as a first-line treatment in secondary stroke prevention.
2. In selected situations, such as erythrocyte alloantibodies or autoantibodies leading to difficult access to transfusions, poor blood availability, or religious objection to blood transfusion, the HU could be indicated as second-line therapy, making clear to the parents that the HU is inferior to chronic transfusions in secondary stroke prevention.
4. Is there evidence that hematologic stem cell transplantation with a matched related donor is a therapeutic option for secondary stroke prevention in patients with sickle cell disease?
Currently, hematologic stem cell transplantation (HSCT) is the only curative treatment for SCD, accepted as a safe and effective therapy. Since the first SCD transplant in 1984, several studies have shown favorable results, mainly with hematopoietic stem cells from compatible sibling donors.40–42(B) Currently, more than 1,250 patients with SCD have been transplanted worldwide. Gluckman et al. published part of these data in 2017, with 1,000 patients between 1986 and 2013 in Europe and in the United States of America. The 5-year overall survival (OS) rate was 92.9% and the 5-year event-free survival (EFS) rate was 91.4%. The cumulative incidence of acute graft versus host disease (aGVHD) grades II and IV was 14.8% and of chronic graft versus host disease (cGVHD), 14.3%.42(B) Recently, Bernaudin et al. published better results in patients with SCD under 30 years old who used myeloablative conditioning (5-year EFS 97.9%, 95%CI 95.5–100%).43(B)
In Brazil, according to ordinance number 1,321, December 21, 2015, allogeneic HSCT with bone marrow or umbilical cord blood from a related donor and myeloablative conditioning is the standard treatment for SCD for several clinical situations, such as stroke, clinically significant neurological events or neurological deficit lasting more than 24 h.
Despite the significant transplant-related mortality and morbidity, the HSCT was able to prevent the occurrence and/or recurrence of stroke and eliminate the need for long-term transfusions.44–47(B)
Walters et al. followed 50 symptomatic patients with SCD (48 HbSS, 1 HbS/O-Arab and 1 HbSβ+), treated with HSCT from a familial HLA-identical donor between 1991 and 1999 when they were under 16 years old. They show relief of pain, no further strokes or ACS and stable neurologic and cognitive functions after the HSCT. Added to this, in 10 engrafted patients with a previous stroke, there was stability or even improvement in radiological images.40(B) Updated results showed that, in a mean follow-up of 3.2 years, 28/29 (97%) patients with previous stroke survived after the HSCT, and 26/29 (90%) survived stroke-free, including stability or improvement in radiological MRI images.48(B)
Bernaudin et al., in 87 SCD (85 HbSS and 2 HbSB0) patients under 22 years old submitted to HSCT between 1988 and 2004, reported no new ischemic lesions after grafting, as well as a reduction in transcranial doppler velocities in 49 patients (from 138 ± 50 cm/s to 100 ± 34 cm/s in the right middle cerebral artery (MCA) and from 138 ± 46 cm/s to 103 ± 40 cm/s in the left MCA). Of note, cerebral vasculopathy was the principal indication for transplantation (55 patients). In this study, 36/87 patients had a previous stroke and only 2 had stroke recurrence: one had a transient ischemic attack on day +10 post-HSCT, while the other, who had previous Moya-Moya disease, progressed to fatal intracranial hemorrhage on day +32 post-HSCT. Regarding vascular occlusions, a resolution was observed in 5 patients, remained unchanged in 16 and the stenoses continued to progress in 2. With a median follow-up of 6 years, the risk of stroke recurrence was 5.6%. Considering that there were no strokes or silent ischemic lesions in patients with successful grafts, including those with previous progressive cerebrovascular disease, the authors suggest that the most critical risk factor for stroke is SCD itself and not the associated vascular disease.49(B)
Results with alternative donors have also been described in recent years, mainly in patients with previous strokes.50,51(B)
1. Clinical question, structured question (PICO) and search strategies
The question structures were made using the PICO system and considering the selected questions.
The articles were identified in the Medline (via PubMed) and manual search (including reference of references, reviews and guidelines).
Initially, the studies were selected by title, sequentially by abstract and, finally, by their full text, which was submitted to critical evaluation and extraction of results related to the outcomes.
Question 1.
Clinical question
Is there evidence that red cell exchange transfusion is the treatment of choice for acute stroke in patients with sickle cell disease?
Structured question (PICO)
Patient: patients with sickle cell disease under 18 years old
Intervention: treatment with red cell exchange transfusion
Comparison: simple transfusion; no treatment
Outcome: secondary stroke prevention
Search strategies
Descriptor: (Anemia, Sickle Cell) AND (Exchange transfusion) AND (Acute Stroke)
Total articles: 97
1st selection: 32
2nd selection: 7
Question 2
Clinical question
Is there evidence that chronic packed red blood cell transfusion can prevent a new episode of stroke (secondary prevention) in patients with sickle cell disease?
Structured question (PICO)
Patient: patients with sickle cell disease under 18 years old
Intervention: chronic blood cell transfusion
Comparison: clinical observation
Outcome: Secondary stroke prevention
Search strategies
Descriptors: (Anemia, Sickle Cell) AND (Secondary Prevention Stroke) AND (Transfusion)
Total articles: 79
1st selection: 40
2nd selection: 17
Question 3.
Clinical question
Is there evidence that hydroxyurea can prevent a new stroke episode (secondary prevention) in patients with sickle cell disease?
Structured question (PICO)
Patient: patients with sickle cell disease under 18 years old
Intervention: treatment with hydroxyurea
Comparison: observation; blood transfusion
Outcome: secondary stroke prevention
Search strategies
Descriptors: (Anemia, Sickle Cell) AND (Hydroxyurea) AND (Stroke)
Total articles: 248
1st selection: 24
2nd selection: 14
Question 4.
Clinical question
Is there evidence that hematologic stem cell transplantation with a matched related donor is a therapeutic option for secondary stroke prevention in patients with sickle cell disease?
Structured question (PICO)
Patient: patients with sickle cell disease, previous stroke and matched related donor
Intervention: hematologic stem cell transplantation
Comparison: clinical observation
Outcome: secondary stroke prevention
Search strategies
Descriptors: (Anemia, sickle cell) AND (Stroke) AND (Transplant)
Total articles: 174
1st selection: 20
2nd selection: 12
2. Initial eligibility criteria for studies
• Components of PICO
• No time limit
• Portuguese, English, French or Spanish
• Full-text availability
3. Selection of articles
Studies were selected independently and blinded after evaluating titles and abstracts of articles obtained using the search strategy, strictly obeying the inclusion and exclusion criteria to separate studies of potential relevance. When the title and abstract were not informative, the article was read in full.
The main reasons for exclusion were that they did not fulfill the PICO criteria and intermediate outcomes. Narrative reviews, case reports, case series and preliminary results were excluded. As the question pertains to treatment, the option was the type of study: comparative observational studies (cohort and/or before and after) and comparative experimental studies (clinical trials).
4. Critical evaluation and strength of evidence
The strength of evidence from observational and experimental studies was defined considering the study design and the corresponding risk of bias, the analysis results (magnitude and precision), the relevance and the applicability (Oxford/GRADE).1,52
The level of Scientific Evidence was classified by the type of study according to the Oxford criteria:1
A: Major experimental and observational studies
B: Minor experimental and observational studies
C: Case reports (non-controlled studies)
D: Opinion without critical evaluation based on consensus, physiological studies or animal models
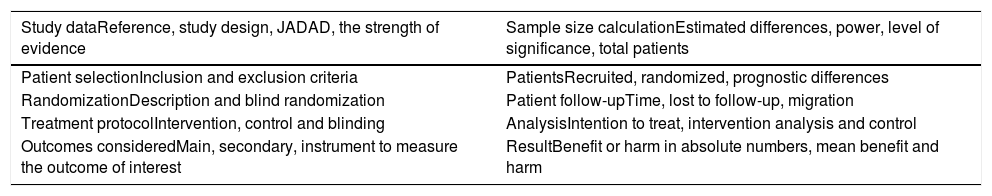
If the selected article in the search was defined as a randomized controlled clinical trial (RCT), it was submitted to an appropriate critical assessment checklist (Table 2). The critical evaluation of the RCT allows classification according to the JADAD score, considering a JADAD score < 3 as inconsistent and articles with a score ≥ 3, consistent.53
Critical outline of randomized controlled trials (checklist).
When the selected article was defined as a comparative study (observational cohorts or non-randomized clinical trials), it was submitted to an appropriate Critical Evaluation Checklist (Table 3), with the classification of the study being according to the Newcastle-Ottawa scale, considering cohort studies consistent with a score ≥ 6 and inconsistent when < 6.54
Scheme of critical evaluation of cohort studies.


