



Acute promyelocytic leukemia has good prognosis in view of the high complete remission and survival rates achieved with therapies containing all-trans retinoic acid or arsenic trioxide. However, there is a significant risk of death during induction due to hemorrhage secondary to disseminated intravascular coagulation. This has contributed to a gap in the prognosis of patients between developed and developing countries. The International Consortium on Acute Promyelocytic Leukemia was created in 2005 and proposed a treatment protocol based on daunorubicin and all-trans retinoic acid stratified by risk geared toward developing countries. Herein are presented the results from the first patient cohort treated in a single developing country hospital employing a slightly modified version of the International Consortium protocol in a real life setting. Twenty patients with acute promyelocytic leukemia were enrolled: 27.8% had low-risk, 55.6% intermediate risk and 16.7% high-risk. The complete remission rate was 94.4% after a median of 42 days. Both relapse rates and death rates were one patient (5.5%) each. No deaths were observed during consolidation. After a median follow-up of 29 months, the overall survival rate was 89.1%. Efficacy and safety of the International Consortium on Acute Promyelocytic Leukemia protocol has been reproduced in acute promyelocytic leukemia patients from a developing country.
Acute promyelocytic leukemia (APL) is a clinical and biological variant of acute myeloid leukemia (AML) characterized by a predominance of abnormal promyelocytes. According to the World Health Organization (WHO), it is currently classified among the AML with recurrent cytogenetic abnormalities [APL and t(15;17)(q22;q12);PML-RARA].1 Untreated, APL is the most aggressive form of AML, with a median survival of under one month.2 It is mainly diagnosed in young adults aged between 20 and 59. Some studies have suggested an increased incidence in Latinos (24.3% vs. 8.3% in non-Latino populations) but this appears to reflect a selection and referral bias.3,4 We recently reported that this type of leukemia accounts for 10.5% of all acute leukemias and 21% of AML at the Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán (INCMNSZ).5
Since the introduction of all-trans retinoic acid (ATRA) in 1988 by Huang in Shangai, complete remission (CR) rates of 85% changed the natural history of this disease.6 The Italian Group for Adult Hematologic Diseases (GIMEMA) reported their experience with regimens including ATRA in combination with anthracyclines, obtaining high CR rates and encouraging disease-free survival (DFS) and overall survival rates (OS).7,8
In 1999, Sanz et al. designed a risk-adapted strategy after observing that patients with high white blood counts (>10×109/L) and low platelet counts (<40×109/L) were at high risk of death during induction and relapse.9 Their study added ATRA to the consolidation regimens in intermediate and high-risk patients. Improvements in treatment have transformed this leukemia into the one with the best survival rates in adults today.
In developed countries, CR rates in APL patients are close to 95% with DFS curves at two years between 87% and 97%.9,10 However, some studies in developing countries reported CR rates of 67.9%, death during induction in 32% of cases and an OS below 60% in 2005.11 The main cause of death during induction was hemorrhage followed by differentiation syndrome.
The current basis of therapy of APL patients hinges on the use of specific agents that induce differentiation, such as ATRA and arsenic trioxide (ATO) in conjunction with chemotherapeutic agents such as anthracyclines.12 ATRA and arsenic trioxide are drugs whose costs are prohibitive to a large group of patients in developing countries.
The International Consortium on Acute Promyelocytic Leukemia (IC-APL) was created in 2005 in order to narrow the gap between the response rates reported in developed countries and those in some developing countries. Its purpose was to integrate an inter-institutional web designed program to generate and implement diagnostic and therapeutic strategies for APL patients in developing countries. This endeavor was supported by expert North American and European work groups. The targeted countries included Brazil, Mexico, Chile and Uruguay. The IC-APL implemented the regimen designed by the Programa para el Estudio de la Terapéutica en Hemopatía Maligna (PETHEMA)/Dutch-Belgian Hemato-Oncology Cooperative Group (HOVON) LPA 2005, in which idarubicin was replaced by daunorubicin (equivalence: daunorubicin 5mg=idarubicin 1mg) in induction and consolidation therapy. This substitution was mainly due to economic limitations. The regimen and its results were recently published.13 CR rates were 85% with a 15% mortality during induction and 5% during consolidation. Median follow-up was 28 months with a relapse rate of 4.5%, OS 80% and DFS 91%.13
The Department of Hematology and Oncology of the INCMNSZ participated in the Consortium's original series with the inclusion of two patients. Because of logistic aspects, we could not include more patients. Due to the encouraging preliminary data at the time, this regimen was later adopted as first-line therapy in APL patients treated at the INCMNSZ as of January 2007. By June 2013, 18 more patients had been treated with this regimen, but not included into the original Consortium study. Patient characteristics, response to treatment and survival are described in this retrospective study of a real-life scenario.
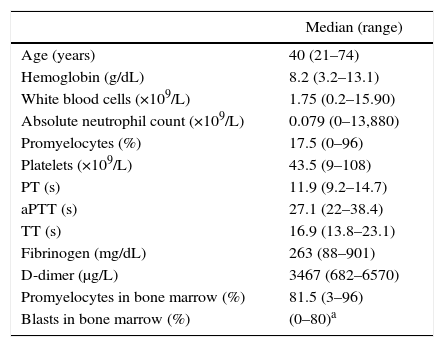
MethodsA retrospective cohort study was carried out of adult patients with APL t(15;17)(q22;q12);PML-RARA, treated in the Acute Leukemia Clinic, Department of Hematology and Oncology of the INCMNSZ, between January 2007 and June 1, 2013. The diagnosis of APL t(15;17) was established according to the WHO criteria.1 Morphological and cytogenetic studies were conducted in all patients (conventional karyotype and FISH with a probe for PML/RARA) in a bone marrow sample obtained at diagnosis. Disseminated intravascular coagulation (DIC) was defined in accordance with the International Society for Thrombosis and Hemostasis criteria (ISTH) if scores were 5 or above.14 Coagulopathy was defined as the prolongation of coagulation times (PT or PTT) or a decrease in fibrinogen that did not fulfill DIC criteria (Table 1).
Baseline characteristics of the patients.
| Median (range) | |
|---|---|
| Age (years) | 40 (21–74) |
| Hemoglobin (g/dL) | 8.2 (3.2–13.1) |
| White blood cells (×109/L) | 1.75 (0.2–15.90) |
| Absolute neutrophil count (×109/L) | 0.079 (0–13,880) |
| Promyelocytes (%) | 17.5 (0–96) |
| Platelets (×109/L) | 43.5 (9–108) |
| PT (s) | 11.9 (9.2–14.7) |
| aPTT (s) | 27.1 (22–38.4) |
| TT (s) | 16.9 (13.8–23.1) |
| Fibrinogen (mg/dL) | 263 (88–901) |
| D-dimer (μg/L) | 3467 (682–6570) |
| Promyelocytes in bone marrow (%) | 81.5 (3–96) |
| Blasts in bone marrow (%) | (0–80)a |
PT: prothrombin time; aPTT: activated partial thromboplastin time; TT: thrombin time.
The induction, consolidation and maintenance regimens were the same as those used by the IC-APL 200613; they were applied without any modification (Figure 1).

During induction, differentiation syndrome prophylaxis was administered with dexamethasone 2.5mg/m2q 12h for two weeks, in patients with a white blood cell count ≥5×109/L. Patients at high-risk and over age 60 were treated as intermediate-risk patients. Maintenance was initiated when hematological recovery was achieved, usually one month after the last consolidation. Support measures during induction and consolidation strictly adhered to the recommendations of the European LeukemiaNet.15
Toxicity was evaluated according to the National Cancer Institute common toxicity criteria scale (version 4.03).16 The only modification to the original protocol was the administration of prophylactic intrathecal chemotherapy to patients considered to be at high-risk; it consisted of methotrexate (12.5mg) and cytarabine (60mg), every month, six dosages, once all three systemic consolidations had been concluded. Modifications were made in monitoring during follow-up – evaluation of minimal residual disease was not performed. At that time, PML/RARA breakpoints were not identified in our center.
Statistics and ethicsThis study was approved by our local Ethics Committee, governed by the principles established in the Declaration of Helsinki for research in humans. Due to the retrospective nature of this report, a consent form was not required; however at our center, we routinely request written authorization for chemotherapy administration and the patient is informed on the risks and benefits. Continuous variables are described as medians and intervals while categorical variables are presented as frequencies and proportions. Between-group differences were established with Mann–Whitney U or Chi square tests for numerical and categorical variables, respectively. Survival curves were created using the Kaplan–Meier method. The SPSS v 15.0 statistical package was used.
ResultsTwenty patients were included but two requested transfers to other hospitals during the diagnostic stage, so 18 patients underwent induction therapy with the modified IC-APL protocol (Figure 2).

Eleven of the 18 evaluated patients (61.1%) were female and seven (38.9%) were male, a ratio of 1.6:1. Median age at diagnosis was 40 with a range of 21–74 years; note that three patients (16.6%) were over 65 years of age at diagnosis. Of the total number of patients, five (27.8%) were classified as low-risk, 10 (55.6%) were intermediate and three (16.6%) were high-risk.
At diagnosis, five patients (27.8%) fulfilled DIC criteria and 38.9% fulfilled those for coagulopathy. None of the patients had abnormal liver or kidney function test results. Of the 18 analyzed patients, nine (50%) had one or more comorbidities: four (22%) had hypothyroidism, two (11%) had type 2 diabetes mellitus, two (11%) were obese, two (11%) had a history of cancer (one patient had had breast and basal cell carcinoma and the other breast cancer), two (11%) had cardiovascular disease (one had systemic hypertension and the other had atrial fibrillation), one (5.5%) had rheumatoid arthritis, one (5.5%) had chronic obstructive pulmonary disease (COPD) and one (5.5%) had relapsing APL and was admitted to the protocol. One of the patients had multiple comorbidities at the time of diagnosis: COPD, obesity, pulmonary tuberculosis, past breast and basal cell carcinoma, post-radiation pulmonary fibrosis, type 2 diabetes mellitus and hypothyroidism; this was the patient that died during induction.
The morphologic characteristics of the bone marrow aspirate were analyzed in 17 cases: 16 (94.1%) had a classic morphology and one case (5.9%) was considered a variant. The diagnosis of AML was initially established in this patient because the variant promyelocytes were counted as blasts. Immunophenotypes were obtained in 16 patients: 100% had a classic immunophenotype: CD34(−), HLA-DR(−), CD13(+), CD33(+), and myeloperoxidase(+). The t(15;17)(q22;q21) translocation was found by karyotype or FISH in 17 patients (94.4%) and only in one case (5.5%) was the demonstration of the PML/RARA rearrangement by polymerase chain reaction necessary.
Of the 18 patients treated with the modified IC-APL 2006 regimen, 17 (94.4%) achieved CR in a median of 42 days (range: 34–158 days). One patient died (5.5%) during induction, the aforementioned patient with multiple comorbidities. Twelve patients (66.7%) developed grade 3–4 infectious complications and two (11.1%) developed grade 3–4 non-hematological complications (subarachnoid hemorrhage and leukocytoclastic vasculitis). The rate of severe febrile neutropenia during induction was 61.1% (11/18) and was more frequent in patients over the age of 65: 100% vs. 53.3%, although this difference was not statistically significant (p-value=0.2). Differentiation syndrome was identified in four patients (22.2%). Interestingly, a group of four patients (22%) did not develop severe complications during induction.
Among the 17 patients in hematologic CR, cytogenetic remission was corroborated by fluorescence in situ hybridization (FISH) in 15; of these, 100% were in complete cytogenetic remission. Molecular remissions were not analyzed.
Of the 17 patients that achieved CR, 14 (82.3%) received three programmed consolidations and three patients (17.6%) received one consolidation one month late; in one case it was due to economic hardship and in two cases, as a result of severe treatment-related complications (severe febrile neutropenia and liver toxicity during the second consolidation). The rate of severe febrile neutropenia during consolidation was 29.4% (5/17). One patient was lost to follow-up after the third consolidation but we corroborated that he was alive and in remission after which we were unable to contact him.
During follow-up, one bone marrow relapse was documented (5.9%). The patient was administered rescue chemotherapy and underwent autologous hematopoietic stem cell transplantation; however, the disease progressed and led to her death. Of all the studied patients, two died (11.1%): one, due to post-chemotherapy aplasia during induction (5.5%) and the other as a result of graft versus host disease (5.5%). At the time of analysis, the treatment protocol had been completed by 10 of the 16 patients in remission (62.5%) and the median overall survival had yet to be reached (Figure 3). After a follow-up of 29 months, OS was 89.1% and only one patient had relapsed (5.8%).
Discussion
To date, this is the first series of APL patients treated with the IC-APL protocol after its publication in 2013. Although the cohort is small, we confirmed the previously published response rates and survival.
As in the IC-APL and European reports, patients at an intermediate-risk prevail (55.5% in the current cohort vs. 52–52.9%) but there was a lower incidence of high-risk patients (16.7% in this cohort vs. 28.17–32%) and a greater incidence of low-risk patients (37.8% in this cohort vs. 16–19.1%).13,17
Our data cannot formally be compared with the previous report of the IC-APL in terms of superiority or inferiority of results because this study was not designed with that goal. Moreover, this study had some limitations such as the small sample size and short follow-up. However, we think that safety aspects and response to treatment were reproduced. Thus, some interesting numbers are worth noting: the CR rate was 94.5% and those described by the original Consortium were 85% and European centers 91–94.3%. In terms of overall survival, it is slightly above that reported by the consortium (89.1% vs. 80% at two years); both were determined after a short follow-up when compared with other reports.13,18,19 Death rates during induction and consolidation were 5.5% and 0%, respectively compared to rates of 15% and 5% reported by the original Consortium.13 These rates are superior to those presented by a Brazilian report before the IC-APL initiative, in which death rates during induction and consolidation were 32% and 10%, respectively.11 Also, in comparison with developed countries, the mortality in the current study is within the previously described range: mortality during induction 5–10% and <5% during consolidation.9,10,17–19
Hemorrhage is the main cause of death described in the literature followed by death due to coagulopathy.13,19 In this study, the rates for both DIC (27.8%) and coagulopathy (38.9%) were low; there were no deaths directly related to hemorrhages.
There are peculiarities to clotting abnormalities and DIC in patients with APL. The incidence of DIC may be possibly overestimated if the diagnosis is established according to the ISTH criteria, since thrombocytopenia and increases in D-dimers score many points in this classification system and they are also common findings in leukemia patients with no other clotting abnormalities.5,14,20 The pathophysiology of fibrinolysis in patients with APL is also different to that of other DIC causes, so these criteria should not necessarily be applied to patients with APL.20 We believe that the value of serum fibrinogen is perhaps the most relevant parameter when defining DIC and for therapeutic decision-making in APL patients; in any case, close dynamic monitoring of these patients is very important during induction.
To date, no central nervous system (CNS) relapses have been observed; however, we cannot reach any solid conclusions on prophylactic treatment of these patients since the sample size was small. CNS prophylaxis with chemotherapy is currently recommended in patients at high-risk of relapse.21
There are many reports on the presence of the FLT3-ITD mutation in APL patients. The frequency of the mutation is variable (12–38%) and its prognostic value is questionable since it has not affected survival curves in all reports.22–25 A recent meta-analysis corroborated that APL patients with the FLT3-ITD mutation have similar CR rates; however, this mutation does lead to shortened OS and DFS due to a greater incidence of relapse.25 The FLT3-ITD mutation has also been correlated to CNS relapse.26 However, to date the presence of the FLT3-ITD mutation has not been incorporated into therapeutic algorithms nor should it be considered in therapeutic decision-making unless it is in the context of a clinical trial.
Although the number of over 65-year-old patients in this cohort is small, they all are nevertheless alive and in CR which coincides with the data reported by other groups that have described long OS in the elderly.27 It is important to mention that the over 65-year olds in this study had severe neutropenia and infectious complications during induction. This underscores the importance of supportive care in this group of patients with APL and in whom curative treatment should always be the goal.
Patients with APL must be treated in specialized centers. Mortality risks, particularly during induction, make them very vulnerable. Although no deaths occurred during consolidation, strict surveillance is also mandatory during this phase. Efforts should focus on the timely prevention and treatment of complications during chemotherapy following the recommendations of expert groups.15
ConclusionsThis is the first cohort of adult patients with APL treated in one center that reproduces the results of the original IC-APL protocol. The CR rates and survival curves observed in this report confirm the effectiveness and safety of this treatment protocol in a real-life scenario.
Conflicts of interestThe authors declare no conflicts of interest.



