



Oxidative stress may aggravate symptoms of hemolytic anemias such as beta-thalassemia. FoxO3 activation results in resistance to oxidative stress in fibroblasts and neuronal cell cultures.
ObjectiveThe purpose of this research was to study FoxO3 gene expression and oxidative status in beta-thalassemia minor individuals.
MethodsSixty-three subjects (42 apparently healthy individuals and 21 with beta-thalassemia minor) were analyzed at the Universidad Nacional de Tucumán, Argentina, between September 2013 and June 2014. A complete blood count, hemoglobin electrophoresis in alkaline pH and hemoglobin A2 levels were quantified. Moreover, thiobarbituric acid reactive species, erythrocyte catalase activity and iron status were evaluated. Beta-thalassemia mutations were determined by real-time polymerase chain reaction. FoxO3 gene expression was investigated by real-time reverse transcription-polymerase chain reaction using mononuclear cells from peripheral blood.
ResultsSubjects were grouped as children (≤12 years), and adult women and men. The analysis of erythrocyte catalase activity/hemoglobin ratio revealed a significant difference (p-value <0.05) between healthy and beta-thalassemia minor adults, but no significant difference was observed in the thiobarbituric acid reactive species levels and FoxO3 gene expression (p-value >0.05). Thiobarbituric acid reactive species and the erythrocyte catalase activity/hemoglobin ratio were not significantly different on comparing the type of beta-thalassemia mutation (β0 or β+) present in carriers.
ConclusionsThe lack of systemic oxidative imbalance demonstrated by thiobarbituric acid reactive species is correlated to the observation of normal FoxO3 gene expression in mononuclear cells of peripheral blood. However, an imbalanced antioxidant state was shown by the erythrocyte catalase activity/hemoglobin ratio in beta-thalassemia minor carriers. It would be necessary to study FoxO3 gene expression in reticulocytes to elucidate the role of FoxO3 in this pathology.
The oxidative status of cells is determined by the balance between pro-oxidants and antioxidants. Oxidants are compounds that can accept electrons, as opposed to reductants that donate electrons. Pro-oxidants are referred to as reactive oxygen species (ROS). The toxicity of ROS depends on their reactivity and life span. Longer life span permits them to diffuse, interact with sensitive biological substrates and cause damage to various organelles. Most of the transition metals, such as iron, can convert relatively stable oxidants into powerful radicals. Excess iron catalyzes hydroxyl radical generation from activated oxygen species by the Haber–Weiss and Fenton reactions.1
The intracellular response to oxidative-stress in erythropoiesis involves the transcription factor, Forkhead box O3a (FoxO3a), which controls pathway(s) regulating erythroid maturation and the levels of oxidative stress in murine erythropoiesis.2 Activation of FoxO3a has been proposed as a protective mechanism in pathological erythropoiesis characterized by abnormal ROS levels such as in β-thalassemia. This FoxO3a effect is mediated by the up-regulated transcription of the ROS scavenging enzymes, superoxide dismutase 2, also known as manganese superoxide dismutase (MnSOD), and catalase.3
Thalassemia is a globin gene disorder that results in a diminished rate of synthesis of one or more of the globin chains and, consequently, a reduced rate of synthesis of hemoglobin (Hb) or Hbs of which that chain constitutes a part. Beta (β)-thalassemia is one of most common autosomal recessive disorders worldwide, and is caused by reduced (β+) or absent (β0) synthesis of the β globin chains of the Hb tetramer, which is made up of two alpha (α) globin and two β globin chains (α2β2).4
A quantitative reduction in β globin results in the accumulation of an excess of α globin chains that are responsible for the pathophysiology of the disorder. The excess of α globin chains forms unstable tetramers that dissociate into monomers and then, followed by a change in their tertiary structure, are oxidized, first to methemoglobin and then to hemichromes which precipitate with time. The following steps are release of hem and free iron (labile iron pool), and precipitation of the protein moiety of the globin, including on the plasma membrane. Cell damage occurs during the production of red blood cell (RBC) precursors in the bone marrow (ineffective erythropoiesis) and in mature RBC in the peripheral blood resulting in shortened life spans. Factors responsible for the oxidative stress in thalassemia are hemoglobin instability and iron in excess.5
β-Thalassemia minor (BTM) is a clinical definition applied to thalassemia patients who present a milder clinical course than those with β-thalassemia major, with a more or less marked anemia that does not require treatment with regular blood transfusions.6 Previous reports were focused exclusively on oxidative stress in patients with β-thalassemia major7,8 and β-thalassemia intermedia.9,10 In severe β-thalassemia, it is difficult to evaluate oxidative stress because of the high proportion of normal red cells due to multiple transfusions. However, oxidative stress has been found to be increased in both β-thalassemia subgroups. In contrast, little is known about the oxidative status in BTM subjects, although Selek et al.6 and Labib et al.11 concluded that oxidative stress is increased. Furthermore, little is known about the relation of oxidative status in BTM subjects with different β-thalassemia mutations. This evaluation is important due to the large genotypic and phenotypic heterogeneity of different populations.
The purpose of this work was to study the FoxO3 gene expression and oxidative status in individuals with BTM, and to correlate it with the type of β-thalassemia mutation (β0 or β+).
MethodsA descriptive cross-sectional study was carried out at the Instituto de Bioquímica Aplicada (Universidad Nacional de Tucumán, Argentina) from September 2013 to June 2014.
SubjectsThis study was approved by Bioethics committee of the Facultad de Medicina (Universidad Nacional de Tucumán), Tucumán, Argentina. Informed consent was obtained from all participants of the study. The sample consisted of 21 BTM subjects (BTM group) and 42 apparently healthy individuals (Healthy group). The inclusion criteria were patients who were diagnosed with BTM and healthy subjects who were older than one year old. Individuals who had ingested vitamins, smokers, diabetics, and patients with coronary heart disease, rheumatoid arthritis, dyslipidemia, hypertension, malignancy, chronic liver disease or renal dysfunction were excluded from the study. Furthermore patients submitted to iron therapy within 21 days prior to analysis, and those who had received transfusions for any reason other than their thalassemia within the three months leading up to the study were not included.
All procedures followed were in accordance with the ethical standards of the committee responsible for human experimentation and with the Helsinki Declaration of 1975, as revised in 2008.
Hematological studiesBlood samples of β-thalassemia carriers were collected in K2-ethylenediaminetetraacetic acid (EDTA). RBC count, mean corpuscular hemoglobin (MCHb) and mean corpuscular volume (MCV) were analyzed with a Sysmex KX-21N hematological counter (Kobe, Japan). Decreased MCV and MCHb allowed an individual to be considered as a potential carrier. The carrier status was confirmed by cellulose acetate membrane electrophoresis at alkaline pH, and the Hb A2 level by microcolumn chromatography (BioSystem S.A., Barcelona, Spain). If Hb A2 was ≥3.5%, then the subjects were diagnosed as β-thalassemia carriers. Serum iron (Fe), total iron binding capacity (TIBC) and transferrin saturation (SAT) were measured by a colorimetric method (Wiener Lab, Rosario, Argentina).
Beta-thalassemia mutations by DNA analysisGenomic DNA was isolated with the High Pure Polymerase Chain Reaction (PCR) Template Preparation Kit (Roche Diagnostics) from 250μL of K2-EDTA anticoagulated whole blood. PCR and mutation detection by melting curve analysis were performed in a LightCycler system 2.0 (Roche Diagnostics), which can simultaneously measure signals emitted from two different fluorophores. PCR primers (forward: 5′-gctgtcatcacttagacctca-3′; reverse: 5′-cacagtgcagctcactcag-3′) were designed to amplify a 587-bp region of the β-globin gene surrounding the most common β-thalassemia mutations in the Argentinian population, as reported previously.12 The clustering of many mutations within small distances permitted the use of two combinations of probes to analyze five common β-thalassemia mutations.
FoxO3 gene expressionThe FoxO3 gene expression was analyzed using real-time reverse transcription-PCR (RT-qPCR). The data obtained from the real time PCR were compared with the expression of the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) gene as an endogenous control (relative quantification). RT-qPCR is a powerful tool to quantify gene expression. The quantitative endpoint for real-time PCR is the threshold cycle (Ct). The Ct is defined as the PCR cycle at which the fluorescent signal of the reporter dye crosses an arbitrarily placed threshold.
RNA was extracted from 1mL of K2-EDTA anticoagulated blood using TRIzol Reagent according to the manufacturer's protocol (Invitrogen). The PCR dissociation curves and subsequent analyses were performed in the LightCycler 2.0 equipment (Roche Diagnostics). The amplicons were subjected to an analysis of ‘melting’ temperature to establish the specificity of the amplified fragments and identity them. The primers and PCR conditions were taken from Kajihara et al.13 The FoxO3 gene expression was calculated using the following formula: FoxO3=2−ΔCt, i.e. [2−(CtFoxO3−CtGAPDH)].
Oxidative stress analysisOxidative damage to biological macromolecules such as DNA, proteins and lipids can be measured by quantifying specific reaction end products. During peroxidation, the peroxides are decomposed to malondialdehyde (MDA), which can be detected by thiobarbituric acid in a colorimetric reaction that produces thiobarbituric acid reactive species (TBARS). The TBARS level was measured spectrophotometrically at 532nm under acidic conditions in plasma obtained after the centrifugation of anticoagulated whole blood.14 TBARS is expressed in μmol/L.
In order to defend themselves against the deleterious effects of ROS, cells maintain an effective antioxidant system consisting of water or lipid-soluble antioxidants and enzymes that remove ROS by metabolic conversion. H2O2 is converted to water and O2 by catalase. Catalase activity differs greatly in diverse human tissues. The highest activity is found in RBC, which is 3600-fold higher than in serum. Thus, the catalase activity of blood is practically all attributable to RBCs.15 Erythrocyte catalase activity (ECAT) was analyzed in K2-EDTA anticoagulated whole blood using the Góth technique with minor modifications to calculate the results.16 The ECAT to blood hemoglobin ratio (ECAT/Hb) was calculated.
Statistical analysisThe Statistical Package for the Social Sciences (SPSS) software version 20.0 was used throughout to carry out all the statistical analyses. Hematological and oxidative stress parameters were compared by the Mann–Whitney U test for pairwise comparisons between groups. The tests were performed with p-value <0.05 as the level of significance. The influence of serum iron on TBARS values and FoxO3 expression, independent of the group (Healthy or BTM), was evaluated by simple regression analysis.
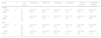
ResultsForty-two healthy subjects and 21 BTM individuals were studied. Table 1 shows the results of hematological parameters, TBARS and the ECAT/Hb ratio in adult groups stratified by gender. The same parameters are observed in Table 2 for healthy and BTM children. As expected, significant differences (p-value <0.05) in hematocrit, hemoglobin, MCV and MCHb were found between BTM and Healthy groups.
Hematological parameters, thiobarbituric acid reactive species (TBARS) and erythrocyte catalase activity (ECAT)/hemoglobin (Hb) ratio in Healthy and beta-thalassemia minor (BTM) adults.
| Group | n | Age [years] | HTOa [L/L] | MCVa [fL] | MCHa [pg] | Hba [g/dL] | TBARS [μmol/L] | ECAT/Hba [MU/g] |
|---|---|---|---|---|---|---|---|---|
| Healthy | ||||||||
| Female (range) | 14 | 34 (13–75) | 0.42b (0.37–0.46) | 88.1 (81.8–96.9) | 29.4 (26.2–31.4) | 11.5b (9.3–14.2) | 0.61 (0.29–0.90) | 0.68 (0.50–1.08) |
| Male (range) | 25 | 37 (14–64) | 0.45 (0.42–0.51) | 86.2 (81.2–93.0) | 29.1 (25.6–31.2) | 14.8 (13.0–17.6) | 0.68 (0.42–0.81) | 0.59 (0.46–0.88) |
| BTM | ||||||||
| Female (range) | 12 | 33 (22–76) | 0.36 (0.33–0.41) | 65.2 (63.0–76.9) | 19.3 (18.3–24.1) | 10.6 (10.0–12.1) | 0.56 (0.38–0.79) | 0.82 (0.66–1.42) |
| Male (range) | 3 | 33 (16–65) | 0.39 (0.32–0.47) | 67.6 (66.9–74.3) | 20.0 (19.8–22.5) | 11.5 (9.3–14.2) | 0.58 (0.40–0.90) | 0.86 (0.76–1.18) |
HTO: hematocrit; MCV: mean corpuscular volume; MCH: mean corpuscular hemoglobin.
Hematological parameters, thiobarbituric acid reactive species (TBARS) and erythrocyte catalase activity (ECAT)/hemoglobin (Hb) ratio in Healthy and beta-thalassemia minor (BTM) children.
| Group (n) | n | Age [years] | HTO [L/L] | MCV [fL] | MCH [pg] | Hb [g/L] | TBARS [μmol/L] | ECAT/Hb [MU/g] |
|---|---|---|---|---|---|---|---|---|
| Healthy (range) | 3 | 11 (8–12) | 0.39a (0.37–0.39) | 81.8a (74.6–84.8) | 26.3a (23.3–28.0) | 122a (121–124) | 0.47 (0.45–0.75) | 0.77 (0.50–0.88) |
| BTM (range) | 5 | 11 (1–12) | 0.35 (0.32–0.38) | 63.0 (58.7–65.1) | 19.2 (17.3–19.5) | 105 (93–112) | 0.51 (0.38–0.58) | 0.83 (0.72–0.90) |
HTO: hematocrit; MCV: mean corpuscular volume; MCH: mean corpuscular hemoglobin.
Lipid peroxidation, as determined by the TBARS level, did not detect significant differences between the Healthy and BTM groups (p-value=0.083). Normal TBARS levels were determined by the 5th and 95th percentiles, and a range from 0.42 to 0.88μmol/L was obtained. Only one BTM subject exceeded the TBARS upper limit.
The comparative analysis of antioxidant status using the ECAT/Hb ratio showed a significant difference (p-value=0.000) between healthy and BTM adults. The normal ECAT/Hb ratio (from 0.50 to 0.91MU/g) was established using the 5th and 95th percentiles.
The FoxO3 gene expression was measured in BTM subjects and in 21 of the 42 healthy subjects; as in the case of TBARS and ECAT/Hb, the 5th and 95th percentiles determined a normal range of from 0.19 to 9.47. Only two women with BTM (9.5%), one with the IVSI-1 G>A mutation and the other with an undetermined mutation, exceeded the upper limit (Figure 1).
; N: Healthy (21 subjects).")
There was no influence of iron levels on TBARS level (R2=0.098) or FoxO3 expression (R2=0.019). Two BTM subjects had low iron (one woman and a 1-year-old child) but this did not impede the observation of an increased Hb A2 level.
The β-thalassemia mutations detected in order of frequency were IVS-I-1 G>A, codon 39 C>T, IVS-I-110 G>A and IVS-I-6 T>C. To evaluate the possible effect of thalassemia mutations on Hb A2, iron status, TBARS, ECAT/Hb ratio and FoxO3 expression, samples were subdivided according to the type of mutation (Table 3). The IVSI-6 T>C (mild phenotype) and IVSI-110 G>A (more severe phenotype) mutations were discriminated in the β+ group. Differences between β0 (11 carriers) and β+ (7 carriers) groups were analyzed, without any significant differences being found (Hb A2: p-value=0.891; Fe: p-value=0.556; TIBC: p-value=0.821; SAT: p-value=0.751; TBARS: p-value=0.079; ECAT/Hb ratio: p-value=0.342; FoxO3 expression: p-value=0.856).
Results of beta-thalassemia minor subjects according to the beta-thalassemia mutations.
| Type of mutationa | n | Hb [g/L] | Hb A2 [%] | Iron [μg/dL] | TIBC [μg/dL] | SAT [%] | TBARS [μmol/L] | ECAT/Hb [MU/g] | FoxO3 gene expression |
|---|---|---|---|---|---|---|---|---|---|
| aβ0 (range) | 11 | 108b (93–121) | 4.8 (4.0–5.6) | 83 (29–259) | 266 (180–347) | 31 (8–97) | 0.60 (0.40–0.90) | 0.92b (0.66–1.42) | 1.84 (0.08–37.01) |
| β+ IVSI-110 G>A (range) | 5 | 106c (100–111) | 4.7 (3.8–5.3) | 80 (75–103) | 297 (220–380) | 32 (20–45) | 0.51 (0.38–0.54) | 0.87 (0.72–1.15) | 2.01 (0.01–7.52) |
| β+ IVSI-6 T>C (range) | 2 | 108 (103–112) | 5.2 (5.1–5.3) | 64 (63–66) | 224 (203–244) | 29 (27–31) | 0.55 (0.52–0.58) | 0.82 (0.81–0.83) | 1.96 (0.53–3.39) |
| Undetermined (range) | 3 | 132 (121–142) | 4.4 (4.2–5.3) | 80 (58–144) | 247 (247–318) | 32 (23–45) | 0.71 (0.48–0.79) | 0.76 (0.73–0.79) | 156.29 (1.75–310.83) |
TIBC: total iron binding capacity; SAT: transferrin saturation; TBARS: thiobarbituric acid reactive species; ECAT/Hb: erythrocyte catalase activity/hemoglobin ratio.
The aim of this study was to investigate the relationship between FoxO3 gene expression and oxidative status in individuals with the β-thalassemia trait. In several studies, an increase of oxidative stress has been reported in both β-thalassemia major and intermedia. The increase of oxidative damage in thalassemia has been related to the generation of free radicals by an excess of denatured α- or β-globin chains, intracellular iron overload and low concentration of normal hemoglobin.7–11
Oxidative status has been investigated in BTM subjects by only a few researchers.6,11,17 All of them showed an increase in oxidative stress and an alteration in antioxidant capacity in β-thalassemia heterozygotes. We recently reported that catalase activity, a key enzyme of the antioxidant system, was increased in only 14% of BTM subjects, with no significant differences between β-thalassemia mutations.16 In order to sharpen the results, the ECAT/Hb ratio was calculated, which was significantly higher in BTM individuals. In the Healthy group, the level of this ratio was similar to that obtained by Vitai and Góth.18 However, Kósa et al. described a decreased ratio between catalase activity in blood and hemoglobin in the BTM group. They proposed that the decrease in catalase activity might be due to the damaging effects of free radicals on the catalase protein.19 However, other authors have reported increased catalase activity in thalassemia individuals.8
Over 200 different mutations have been found to cause β-thalassemia, with splicing mutations among the most common. Most of these mutations activate aberrant cryptic 5′-donor or 3′-acceptor splice sites without completely abolishing normal splicing. These mutations lead to the production of variable amounts of normal transcripts. Some mutations allow a significant level of normal splicing (such as IVSI-6 T>C), leading to thalassemia intermedia, whereas others reduce normal splicing to low levels (such as IVSI-110 G>A) or very low levels (such as IVSI-5 G>A and IVSII-654 C>T), causing a transfusion-dependent disease in homozygotes.20 Therefore, IVS-I-110 G>A, although considered β+, is a very severe genotype with a minimal amount of Hb A (generally, less than 5%). So the lack of any significant difference between β0 and β+ in the present study could be related to the fact that only two patients are genuinely β+ (IVS-I-6 T>C).
In respect to increased oxidative stress, our findings were not consistent with previous studies. The oxidant status of BTM subjects showed no significant differences with the Healthy group. Selek et al., by measuring the total oxidant status (TOS), total antioxidant capacity (TAC), lipid hydroperoxide (LOOH) levels and oxidative stress index (OSI), detected an increase of oxidative status.6 Furthermore, Ondei et al., by analyzing TBARS and Trolox equivalent to antioxidant capacity (TEAC), identified an increment of oxidative stress and antioxidant capacity in β-thalassemia heterozygotes, mainly in carriers of the codon 39 mutation.17 Labib et al. measured TAC and TBARS. They found that TAC was significantly lower in BTM subjects than in controls, while TBARS was significantly higher, with significant differences between the β0 and β+ thalassemia traits.11 In addition, Lai et al. reported higher levels of the pro-oxidant/antioxidant ratio in β-thalassemia intermedia and BTM, compared to controls.21 Differences with the present study may be due to methodological differences and sensitivity of the methods.
In this study, there was no relationship between serum iron, TBARS and FoxO3 expression. In accordance, Ondei et al. did not detect an influence of iron levels, SAT or ferritin on TBARS and TEAC values.17 Al-Hakeim et al. reported no statistically significant correlation between iron status parameters and MDA or total carbonyl in patients with β-thalassemia major.22 Excessive iron concentrations can stimulate lipid peroxidation in thalassemia mouse models, which resemble thalassemia patients receiving blood transfusions,23 however these studies have indicated no direct association between serum iron parameters and MDA.22,23 Instead, the strongest predictor of increased MDA in patients with severe thalassemia would be the liver iron concentration.24
The mammalian forkhead transcription factors of the O class (FoxOs) has four members: FoxO1, FoxO3, FoxO4, and FoxO6. FoxO1 and FoxO3 are expressed in nearly all tissues. FoxOs transcriptionally activate or inhibit downstream target genes, therefore playing an important role in proliferation, apoptosis, autophagy, metabolism, inflammation, differentiation, and stress resistance. FoxO1 and FoxO3 play an important role in protection of cells against oxidative stress.25 Wang et al. found that oxidative stress enhances the expression of FoxO3a but not FoxO1 and FoxO4 in endothelial progenitor cells.26 FoxO3 increases the levels of MnSOD in mitochondria, which removes superoxide radicals. In vivo experiments suggest that FoxO3 protects against oxidative stress by increasing MnSOD expression and production of catalase and peroxiredoxin III.27 FoxO3 is also critical for hematopoietic self-renewal. In vivo experiments have established that FoxO3 deletion in hematopoietic stem cells increases ROS and impairs their hematopoietic capacity.28 FoxO3 also protects erythropoiesis against oxidative stress.2 Resveratrol, a novel potential therapy that ameliorates oxidative stress in circulating β-thalassemia red cells, activates FoxO3 transcriptional factor.29 The use of rapamycin, a mechanistic target of rapamycin (mTOR) inhibitor, has been tested in β-thalassemia mice and remarkably improved erythroid cell maturation, β-globin production and anemia through FoxO3 activation.30 Therefore, up-regulation of FoxO3 could be a target to control oxidative stress in β-thalassemia subjects.31
Our data show that FoxO3 gene activation was increased in only two BTM individuals. FoxO3 gene expression was normal in most BTM subjects, and this observation could be related to the lack of oxidative imbalance shown by TBARS levels in plasma. Although FoxO3 expression in peripheral mononuclear cells is not strictly representative of oxidative stress in the erythroid lineage, their assessment is valid because both cell types interact in the same environment.
A limitation of this work is that superoxide dismutase was not measured; this could have contributed to the evaluation of the antioxidant status of the carriers. The low number of samples is another limitation of this study.
ConclusionsOxidative stress plays an important role in the pathophysiology of β-thalassemia, and its control could ameliorate the clinical signs of the disease. FoxO transcription proteins modulate several cellular functions through regulation of multiple transcriptional targets. Therefore, regulation of FoxO transcription factors by antagonists in some disease states, or agonists in normal and disease conditions, may be useful in treating or preventing a wide variety of disorders. In the present work, the presence of systemic oxidative stress was not proved in BTM individuals, and in most, the FoxO3 gene expression was not up-regulated. However, the study of antioxidant activity in RBC demonstrated an increased ECAT/Hb ratio. An analysis of reticulocytes separated from the blood of BTM subjects may clarify the role of FoxO3 in this condition. Furthermore, it would be necessary to perform a cohort study of β-thalassemia heterozygotes over a long period of time to recognize their oxidant status and its relation with the type of mutation.
FundingThis work was realized with grants granted by the Consejo de Investigaciones de la Universidad Nacional de Tucumán (CIUNT 26/D520).
Conflicts of interestThe authors declare no conflicts of interest.
This work counted on the support of the Consejo de Investigaciones de la Universidad Nacional de Tucumán (CIUNT 26/D520), who granted the funds for the research. The authors thank Biochemistry Specialist Guillermo Fabián Vechetti and the Laboratorio Tucumán for the use of its molecular biology equipment.



