



This study aimed to describe and analyze clinical and laboratory characteristics of patients with sickle cell anemia treated at the Hemominas Foundation, in Divinópolis, Brazil. Furthermore, this study aimed to compare the clinical and laboratory outcomes of the group of patients treated with hydroxyurea with those patients that were not treated with hydroxyurea.
MethodsClinical and laboratorial data were obtained by analyzing medical records of patients with sickle cell anemia.
ResultsData from the medical records of 50 patients were analyzed. Most of the patients were female (56%), aged between 20 and 29 years old. Infections, transfusions, cholecystectomy, splenectomy and systemic arterial hypertension were the most common clinical adverse events of the patients. The most frequent cause of hospitalization was painful crisis. The majority of patients had reduced values of hemoglobin and hematocrit (8.55±1.33g/dL and 25.7±4.4%, respectively) and increased fetal hemoglobin levels (12±7%). None of the clinical variables was statistically significant on comparing the two groups of patients. Among hematological variables only hemoglobin and hematocrit levels were statistically different between patients treated with hydroxyurea and untreated patients (p-value=0.005 and p-value=0.001, respectively).
ConclusionSickle cell anemia requires treatment and follow-up by a multiprofessional team. A current therapeutic option is hydroxyurea. This drug reduces complications and improves laboratorial parameters of patients. In this study, the use of the drug increased the hemoglobin and hematocrit levels of patients.
Sickle cell anemia (SCA) is an inherited autosomal recessive disease characterized by the presence of homozygous hemoglobin S (Hb S). It is caused by a single nucleotide mutation that substitutes glutamic acid for valine in the sixth position of the ¿-globin gene.1 During hypoxic conditions, the red blood cell becomes sickled and the resulting change in structure restricts circulation causing obstruction of the blood flow within the capillaries and early destruction of the cell.2
Clinical manifestations of SCA vary from mild, that is, almost asymptomatic, to severe forms that are associated with high mortality rates.3 Clinical manifestations usually appear after three months of age, when the concentration of fetal hemoglobin (Hb F) decreases.2 Most systems are liable to vaso-occlusive processes possibly resulting in multisystem failure.4–6
A definitive cure is not currently available for patients with SCA. Existing therapies are only focused on symptom management and do not alter the natural history of the disease. These therapies are comprised of hydration, prevention of infections, pain management, proper nutrition and precautions against adverse weather conditions. Thus, additional therapies are needed to prevent complications without subjecting patients to the increased morbidity and mortality associated with highly aggressive approaches such as hematopoietic stem cell transplantation (HSCT).
Currently, hydroxyurea (HU) is the only medical modality with proven efficacy in patients with frequent symptoms related to SCA.7,8 HU is known to increase Hb F levels, improve hemoglobin concentrations and mean corpuscular volume, and reduce the number of reticulocytes. Another favorable response of treatment is that it does not only reduce the expression of adhesion molecules, but also decreases the number of receptor proteins located on endothelial cells. Therefore, HU decreases vascular adhesion which contributes by diminishing the number of vaso-occlusive crises.9,10
SCA is an inherited disease with high prevalence and mortality rate.11 However, the literature is scarce on local epidemiological studies in Brazil. Hence, this study aims to analyze the clinical and laboratorial characteristics of patients with SCA who live in the macro region of Divinópolis, Minas Gerais treated in the Hemominas Foundation. Furthermore, this study aims to compare the clinical and laboratorial outcomes in two groups of patients; those treated with HU to those that were not treated with HU.
MethodsStudy sample and data collection procedureA retrospective study was carried out based on information extracted from medical records. This study was conducted at the blood center of the Hemominas Foundation in Divinópolis, Brazil. All patients from that macro region diagnosed with SCA (limited to the homozygous Hb SS genotype) and followed-up from August 2012 to August 2014 were included. Initially the planned sample was composed of 57 patients that is, all cases of SCA at the blood center. However, seven patients were excluded from analysis (five were not followed-up in the previous two years, one patient died and one patient was transferred to another blood center).
This study was approved by the Research Ethics Committee of the Universidade Federal de São João Del Rei, Campus Centro Oeste Dona Lindu (# 477.473) and by the Research Ethics Committee of the Hemominas Foundation (# 506.674).
A standardized data extraction form was used to collect information from patients’ medical records. All data collected refer to the period of interest (August 2012 to August 2014). The form contained the following information: age, date of diagnosis, date of starting treatment, adherence to the institutional vaccination protocol (protection against influenza, meningococcus and pneumococcus), clinical and therapeutic aspects, and laboratory tests results. The arithmetic mean was calculated from the last five laboratory test results within the period of the trial.
Statistical analysisDescriptive statistics are used to report the variables of interest. Categorical variables are reported as absolute and relative frequencies. Continuous variables are presented as means and standard deviation or median and interquartile range if the variable showed non-parametric distribution. Distribution of the data was tested using the Shapiro–Wilk test. Normally distributed data were analyzed using the Student's t-test, whilst non-parametric data were analyzed using the Mann–Whitney U test. Pearson's chi-square test or Fisher's exact test, as appropriate, were used to associate exposure with outcomes. These analyses were performed using the Statistical Package for Social Sciences (SPSS Inc., Chicago) version 22 for Windows. The level of significance was set at 5% (p-value <0.05).
ResultsData from the medical records of 50 patients were analyzed. Females, with a total of 28 (56%), were predominant in the sample. The age range of the patients was 2–54 years with a mean of 25.4±12.9 years. When grouped by age, the majority of the patients were between 20 and 29 years (36%) while the over 50-year-old age group was the smallest (4%).
The age at diagnosis was reported in 30 patient records, with 14 (46.7%) patients diagnosed at less than one year of age (median: 1.0; interquartile range: 0.0–10.5 years) as part of the Newborn Screening Program in Minas Gerais. Regarding the number of medical appointments over the previous two years, 50% of patients had 17 medical appointments or more (interquartile range: 11–32 consultations).
Clinical characteristicsSeven patients (14%) underwent splenectomy and 15 patients (30%) were submitted to cholecystectomy. Nine of the 50 patients had systemic arterial hypertension (18%). Splenic sequestration was not mentioned in any of the records within the period studied. The vaccination protocol was described in 31 records with 24 patients (77.4%) adhering to the protocol established by the Brazilian Health Ministry12; all under 20-year-old patients adhered to the vaccination protocol. During the study period, 20 patients (40%) received blood transfusions. Previous infectious events were recorded for 26 patients (52%), with 65.4% having just one infectious episode. The predominant cause was upper respiratory tract infection (46%), in particular tonsillitis (20% of infections). Twenty-one patients (42%) were hospitalized mainly for painful crises in 43.3% of all cases (Table 1).
Clinical characteristics of 50 patients with sickle cell anemia.
| Characteristic | n | % |
|---|---|---|
| Splenectomy | 7 | 14.0 |
| Cholecystectomy | 15 | 30.0 |
| Systemic arterial hypertension | 9 | 18.0 |
| Chronic renal failure | 3 | 6.0 |
| Vaccination protocol (n=31) | 24 | 77.4 |
| Transfusions | 20 | 40.0 |
| Infections | 26 | 52.0 |
| 1 event | 17 | 65.4 |
| ≥2 events | 9 | 34.6 |
| Hospitalization | 21 | 42.0 |
| 1 event | 16 | 76.2 |
| ≥2 events | 5 | 23.8 |
| Priapism (n=22) | 5 | 22.7 |
| Painful crisis | 30 | 60.0 |
| Others | 7 | 14.0 |
| Referrals | ||
| Cardiology | 32 | 64.0 |
| Ophthalmology | 38 | 76.0 |
| Neurology | 9 | 18.0 |
| Other medical specialties | 23 | 46.0 |
| Medications | ||
| Hydroxyurea | 15 | 30.0 |
| Vitamins | 50 | 100.0 |
| Analgesic/anti-inflammatory | 26 | 52.0 |
| Iron-chelating | 11 | 22.0 |
| Antibiotic | 9 | 18.0 |
| Other | 16 | 32.0 |
Others: stroke, chronic hypoxemia, acute chest syndrome, bone necrosis, proliferative retinopathy, hematuria.
Regarding common acute events in SCA, priapism was reported in five (22.7%) men and painful crisis in 30 (60%) patients (Table 1). Patients were referred for medical specialist appointments for a variety of reasons, with the greatest number of referrals being made to ophthalmology (76%) and cardiology (64%) services (Table 1).
Pharmacological therapiesHU was prescribed to 15 patients (30%) at the recommended initial single dose for adults of 15mg/kg/day.9,12 Indications for HU treatment included age older than three, history of vaso-occlusive crises that required medical support, recurrent acute thoracic crises, strokes, recurrent priapism and severe persistent anemia in the previous 12 months.13 Even though six patients had a clinical indication for HU treatment, they did not start treatment. In four cases, this was because the patients were unable to attend frequent medical appointments and therefore could not perform periodic laboratorial tests required as per the protocol.9,12 Of the other two patients, one did not receive HU as she was trying to conceive, and one was a child who did not have an adult to take responsibility for his treatment. All patients took some vitamins, such as folic acid, and analgesic/anti-inflammatory drugs were widely used (52% – Table 1).
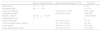
Laboratorial characteristicsThe descriptive analysis of hematological variables is shown in Table 2. The hematocrit, total hemoglobin, fetal hemoglobin, ferritin and lactate dehydrogenase values are adjusted taking into account the reference values for both gender and age group (Table 2).
Description of hematological variables of 50 patients with sickle cell anemia.
| Mean (± standard deviation) | Median (interquartile range 25–75%) | Referencea | |
|---|---|---|---|
| Hematocrit (%) | 25.7±4.4 | – | 34–49 |
| Hemoglobin (g/dL) | 8.55±1.33 | – | 11.5–17.5 |
| Leukocytes (cell/mm3) | – | 10.354 (8.271–13.846) | 3500–12,000 |
| Neutrophil (cell/mm3) | – | 4.945 (3.664–6.914) | 1500–8000 |
| Platelets (cell/mm3) | 433.418±134.348 | – | 15,000–400,000 |
| Fetal hemoglobin (%) | 12±7 | – | 0 |
| Serum iron (¿g/dL) | – | 129.3 (86.0–174.0) | 35–160 |
| Total iron-binding capacity (mg/dL) | – | 307.5 (276.5–361.0) | 228–428 |
| Ferritin (ng/mL) | – | 344.0 (152.3–771.0) | 10.0–300.0 |
| Lactate dehydrogenase (UI/L) | – | 752.0 (541.9–913.0) | 150.0–400.0 |
Table 3 compares the incidence of clinical adverse events between the groups that received HU to those that did not. There was no statistically significant difference for the most prevalent clinical events. The incidence of the category ‘others’, which includes the less common events such as stroke, acute chest syndrome, chronic hypoxemia, bone necrosis, proliferative retinopathy and hematuria, was statistically different between the two groups of patients with these events being more prevalent in individuals that took HU (Table 3).
Clinical characteristics of 50 patients with sickle cell anemia treated (on HU) and not treated (off HU) with hydroxyurea.
| Off HU (n=35) | On HU (n=15) | p-Value | |
|---|---|---|---|
| Systemic arterial hypertension | 8 (22.9%) | 1 (6.7%) | 0.247 |
| Splenectomy | 4 (11.4%) | 3 (20.0%) | 0.415 |
| Cholecystectomy | 9 (25.7%) | 6 (40%) | 0.333 |
| Vaccination protocol (n=31) | 18 (78.3%) | 6 (75.0%) | 1.000 |
| Transfusions | 13 (37.1%) | 7 (46.7%) | 0.529 |
| Infections | 19 (54.3%) | 7 (46.7%) | 0.621 |
| Hospitalization | 13 (37.1%) | 8 (53.3%) | 0.288 |
| Priapism (n=22) | 2 (13.3%) | 3 (42.9%) | 0.274 |
| Painful crisis | 20 (57.1%) | 10 (66.7%) | 0.529 |
| Others | 2 (5.7%) | 5 (33.3%) | 0.020a |
Others: stroke, chronic hypoxemia, acute chest syndrome, bone necrosis, proliferative retinopathy, hematuria.
Table 4 shows a descriptive and comparative analysis of hematological variables between the two groups. Hematocrit and hemoglobin levels were significantly different (p-value 0.005 and 0.001, respectively). There were no significant differences between the groups in respect to the other variables (Table 4).
Description of hematological variables of 50 patients with sickle cell anemia treated (on HU) and not treated (off HU) with hydroxyurea.
| Off HU (n=35) | On HU (n=15) | p-Value | |
|---|---|---|---|
| Hematocrit (%)a | 24.6±4.1 | 28.3±4.1 | 0.005c |
| Hemoglobin (g/dL)a | 8.15±1.22 | 9.44±1.14 | 0.001c |
| Leukocytes (cell/mm3)b | 10,502 (8361–14,776) | 9370 (7518–13,600) | 0.121 |
| Neutrophil (cell/mm3)b | 5118 (4000–7005) | 3760 (3016–6689) | 0.494 |
| Platelets (cell/mm3)a | 431,541±149,615 | 437,547±96,969 | 0.869 |
| Fetal hemoglobin (%)a | 11±5 | 14±9 | 0.212 |
| Serum iron (¿g/dL)b | 127.5 (80.2–171.3) | 151.5 (106.7–175.0) | 0.438 |
| Total iron-binding capacity (mg/dL)b | 313.3 (277.3–368.0) | 296.0 (273.0–352.5) | 0.398 |
| Ferritin (ng/mL)b | 326.0 (114.0–677.6) | 528.5 (250.0–1240.2) | 0.201 |
| Lactate dehydrogenase (UI/L)b | 752.0 (663.5–958.0) | 781.5 (438.5–897.7) | 0.557 |
HU: hydroxyurea.
According to the results of this study, most patients were female, predominantly aged between 20 and 29 years of age and had been diagnosed at around the age of one. Similar findings were reported in other studies from Brazil; the majority of cases were women and were aged between 18 and 30 years.14 One study conducted in Paraná showed a predominance of females with a gender ratio of 2:1 in children with SCA.15 In a national study of patients with sickle cell disease, the average age at diagnosis was 8.4 years.14 In this study, the median age at diagnosis was one year, which meets the current national expectations set by the National Program of Neonatal Screening by the Brazilian Health Ministry in 2001.
Regarding the clinical characteristics, the rate of cholecystectomies was high (30%) in this sample compared to the general population. This number is due to the greater risk of these individuals developing gallstones, which is in accordance with what has been previously reported in the literature, estimated at 50%.16,17 Splenectomy was another procedure commonly performed in the patients of the current study (14%). This surgical procedure is aimed at reducing deaths from recurrent episodes of acute splenic sequestration; it sometimes improves nutritional status and hematological levels related to chronic hypersplenism. According to the literature, the prevalence of acute splenic sequestration is estimated at around 7.5–30%.18,19 Although not all patients investigated in this study were referred for surgery, the rate of splenectomy in this study was within the expected range.
Nine of the 50 patients studied had systemic arterial hypertension (18%). Perhaps this finding is due to the high prevalence of this disease among Brazilian adults in general (20%).20 Although experimental and clinical studies report the impact of nitric oxide depletion on the pathogenesis of pulmonary hypertension in SCA, no studies correlate arterial hypertension to SCA.21
There was a high rate of infections in this sample (52%). These findings can be explained by the fact that autosplenectomy generally occurs before the patients are 5 years old. Autosplenectomy is caused by splenic infarcts culminating in atrophy and fibrosis of the spleen. Even prior to this process, splenic lesions can lead to functional asplenia, which can become permanent when the patient has reached 6–8 years of age, making them liable to infections. In fact, infections have been reported as one of the most frequent complications in individuals with SCA.2
Most patients (77.4%) were submitted to the vaccination protocol. The literature explains this finding by the early diagnosis of SCA and screening programs, which enables the follow-up of children with SCA with early antibiotic prophylaxis and immunization against encapsulated bacteria.2
Painful crises in 42% of patients were the commonest reason for hospitalization during the two years of this study. Pain, the most impacting presentation of the disease, is the result of obstruction of the microcirculation caused by sickled red blood cells. It frequently happens without prodromes and affects the patients’ quality of life.22
Ophthalmology and cardiology diseases were the two most common motives for patient referrals (76% and 64% of patients, respectively). Previous studies have shown the importance of ophthalmic evaluations in patients with SCA in order to prevent advanced eye disorders.23 Cardiomegaly, myocardial ischemia, biventricular dysfunction and pulmonary hypertension are the most frequent clinical complications related to cardiovascular changes in sickle cell disease. They may occur due to chronic hemolytic anemia, or secondary to pulmonary disorders or to iron overload from blood transfusions.24
The literature shows favorable outcomes with the use of HU, such as increases in hemoglobin, mean corpuscular volume, number of leukocytes, neutrophils and platelets.25,26 On comparing the two groups, increases in hemoglobin and hematocrit values were statistically significant in this study.
There were no significant differences between the patients receiving HU compared to those who were not being treated with HU with regard to painful crises, infections, hospitalizations and number of transfusions, unlike other studies that indicated an improvement in these clinical outcomes.19 However, it is important to highlight some points that could contribute to this difference, particularly in relation to clinical and laboratory outcomes. By definition, patients who required HU were at a more severe stage of the disease as they met the clinical criteria for the use of HU. In contrast, the group that did not receive HU was comprised of clinically heterogeneous patients. Perhaps the patients who did not use the medication presented with a milder form of the disease and therefore, did not meet the criteria for using the medication. Despite this, six patients had clinical indications to start treatment with HU but did not as they did not attend medical appointments frequently enough as demanded by the protocol. Thus, the group not receiving the medication was comprised of patients with varied clinical presentations. A comparison between a group with clinical indications that is taking HU and a group without clinical indications who did not take HU would provide a more accurate idea of the benefits of the drug. However, methodological issues such as the total number of patients and the cross-sectional design of the study preclude definitive conclusions.
This study has some limitations that need to be acknowledged. The population investigated was very specific and was not randomly selected. Therefore, this sample may not be representative of the population, affecting the generalization of the results. Furthermore, the heterogeneity of patients, and the relatively small sample size presented a challenge since it was not possible to perform sub-group analyses. Future studies should investigate a larger sample to overcome this limitation. Additionally, the patients’ medical records could not be linked to records from other healthcare facilities, such as hospitals. In summary, more studies are needed, particularly those that focus on grouping patients with similar clinical presentations prior to comparison.
ConclusionSickle cell anemia requires treatment and follow-up by a multiprofessional team. A current therapeutic option is hydroxyurea. This drug reduces complications and improves laboratorial parameters of patients. In this study, the use of the drug increased the hemoglobin and hematocrit levels of patients.
Conflicts of interestThe authors declare no conflicts of interest.
We acknowledge the support by Universidade Federal de São João Del Rei (UFSJ), Programa Institucional de Bolsas de Iniciação PIBIC, Fundação de Amparo à Pesquisa do estado de Minas Gerais (FAPEMIG), Bolsa de Iniciação Científica PIBIC; Conselho Nacional de Desenvolvimento Científico e Tecnológico – CNPq/Brazil (Processo: 442189/2014-1); and Fundação Hemominas – Hemonúcleo Regional de Divinópolis, in the city of Divinópolis, Minas Gerais, Brazil.



