



Paroxysmal nocturnal hemoglobinuria is an acquired chronic hemolytic anemia, which often manifests as peripheral blood cytopenias and thrombosis.
ObjectiveThe aim of this study is to describe a Brazilian population of paroxysmal nocturnal hemoglobinuria patients.
MethodsOne hundred and three paroxysmal nocturnal hemoglobinuria cases were retrospectively reviewed and the clinical presentation, thrombosis, survival, and clone size were assessed. Diagnosis was established by flow cytometry.
ResultsFifty-two male and 51 female patients with a median age of 24.1 years (5.5–62 years) were studied. Clinical symptoms included hemoglobinuria (18.4%), infection (46.6%) and thrombosis (16.5%), and 80.6% had pancytopenia. Patients were classified as classic paroxysmal nocturnal hemoglobinuria (10), paroxysmal nocturnal hemoglobinuria with aplastic anemia (39), and paroxysmal nocturnal hemoglobinuria with subclinical features and aplastic anemia (54). There were significant differences in terms of median age, size of clone, clinical symptoms, and peripheral blood cell counts between the three subcategories. The clone size in erythrocytes and granulocytes were respectively 0.04% (range: 0–18%) and 7.3% (range: 0.3–68.7%) in patients with subclinical features and aplastic anemia, 15.8% (range: 0–99.7%) and 63.0% (range: 1.7–99.8%) in patients with aplastic anemia alone, and 82.2% (range: 0–99.85%) and 98.0% (81.3–100.0%) in Classic disease. Statistical differences were identified for platelets (p-value=0.001), lactate dehydrogenase (p-value=0.002) and the clone size (p-value<0.001) in patients who suffered thrombotic events compared to those who did not. Overall survival was 81.7%, with patients with subclinical features and aplastic anemia having lower overall survival (76.5%).
ConclusionThis retrospective review of 103 patients over an 11-year period represents the largest collection of paroxysmal nocturnal hemoglobinuria cases from a single center in Brazil. Flow cytometry showed that a larger clone was associated with classical symptoms and increased risk of thrombosis, even in patients with bone marrow failure, whereas a smaller clone was associated with bone marrow aplasia.
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare, acquired, stem cell disorder characterized by hemolytic anemia, bone marrow failure, and an acquired thrombophilic state.1–4 Manifestations of the disease are related to complement-mediated intravascular hemolysis due to the lack of glycosyl phosphatidylinositol-anchored complement regulatory proteins (GPI-AP), CD55 and CD59 on red blood cells.5,6 Patients with PNH may present not only with a wide range of clinical manifestations such as weakness, pallor, and asthenia due to hemolysis, but also abdominal pain, dysphagia, or pulmonary hypertension.2–4 Thrombosis, often occurring at unusual sites, is a major life-threatening risk for patients with PNH.1–4 Ten-year risk of thrombosis has been associated with the PNH clone, as patients with large PNH clones (>50%) had 44% of 10-year risk compared with 5.8% in patients with small clones.7 A frequent association between PNH and aplastic anemia (AA) has been described, with two potential patterns of evolution: progressive marrow failure in patients without detectable PNH clones or AA in patients in whom a PNH clone is detected.8–10
Diagnosis of PNH has improved over the years with the adoption of modern technologies. Two decades ago the Ham test, which is based on increased sensitivity of PNH-affected red blood cells to complement-mediated lysis, was used.5,11 Today, the Ham test has usually been substituted by the more sensitive, informative and less cumbersome flow cytometric assay (FCM), which uses antibodies directed against the GPI-AP.11–14 PNH cells are characterized by GPI-AP deficiency on the cell surface due to an acquired mutation of the phosphatidylinositol glycan-class A (PIGA) gene in one or more hematopoietic stem cells.6,11,12 The development of FCM-based testing has allowed the detection of small PNH clones, which would otherwise not be evident.13,15 PNH clones are also detected in the setting of bone marrow failure, and about 40–50% of AA patients have a PNH clone detected at the time of diagnosis.8,15,16 The mechanism by which the expansion of PNH cells occurs in AA remains unclear; one hypothesis is that PNH cells have a proliferative advantage over non-PNH cells by an immune selection mechanism.9,10 The presence of a PNH clone has been reported to be predictive of a response to immune suppression in AA15 but other authors did not observe this finding.16 For optimum management, the contribution of both hemolysis and marrow failure to the complex anemia of PNH should be determined.1
The objective of this study was to assess the clinical presentation of PNH patients at the time of diagnosis, as well as report complications, such as thrombosis, survival, difference between subcategories and clinical significance of the PNH clone size. Patients were assigned to one of the three subcategories, namely Classic PNH, PNH/AA syndrome, and subclinical PNH (PNH-sc/AA) to explore the differences between these categories. Furthermore, the size of PNH clone was evaluated in the entire cohort and in each subcategory to assess whether the size of PNH clones was associated with some of the clinical features of PNH.
MethodsPatients and study designOne hundred and three PNH clone cases referred to a tertiary medical center in Brazil from December 1999 through December 2011 were retrospectively reviewed. A total of 398 patients were screened for the PNH clone using a FCM assay.5,14 The diagnosis of PNH was established by detecting a GPI-AP deficient clone greater than 0.1%, with at least two cell lineages showing GPI deficient populations. The study included 103 patients who had demonstrated the presence of a PNH clone and had available clinical data. The date of PNH diagnosis was based on the first positive FCM analysis. Patients with co-morbid AA were subclassified as severe or non-severe according to published criteria.17,18
Multiparameter flow cytometryThe diagnosis of PNH was established by the detection of an unequivocal positive PNH clone by multicolor FCM assay5,11 using a FACSCalibur® cytometer (BD Biosciences, San Jose, USA) and CellQuest Pro software (BD Biosciences, San Jose, USA). The proteins studied were CD55 and CD59 on red cells, neutrophils and monocytes; CD16, CD24 and CD66b on neutrophils and CD14 on monocytes. PNH clones were defined by the presence of GPI-AP deficient cells at a frequency greater than 0.1% of neutrophils, monocytes and red cells, and the proportion of GPI-AP deficient cells (clone size) was defined by the highest level of these cells lacking GPI-anchored proteins.
Debris was thresholded out, and at least 50,000 events in leukocyte tubes and 20,000 events in red cell tubes were collected and analyzed using Paint-a-gate® (BD Biosciences, San Jose, USA) or Infinicyt™ (Cytognos, Salamanca, Spain) software (for samples tested after January 2009). Red cells and granulocytes were identified based on forward and side scatter, and by staining with CD41a FITC and CD45 PercP, respectively. The gates used to define GPI negative populations were established by using normal red cells and granulocytes as controls.
Subcategories of paroxysmal nocturnal hemoglobinuria patients at diagnosisPatients were assigned to one of the three subcategories based on the recently proposed PNH working clinical classification1:
- (I)
The Classic PNH subcategory included patients with clinical and laboratory evidence of intravascular hemolysis (such as hemoglobinuria, hemoglobinemia, and elevated LDH and bilirubin) but no evidence of bone marrow failure;
- (II)
Patients in the PNH/AA subcategory were defined by the presence or a history of bone marrow failure in conjunction with clinical and laboratory evidence of intravascular hemolysis;
- (III)
Patients in the subclinical PNH (PNH-sc/AA) subcategory were identified by the presence of bone marrow failure but without clinical or laboratory evidence of hemolysis.
The criteria for bone marrow failure included bone marrow hypoplasia (cellularity<50%) and at least two of the following three laboratory abnormalities: hemoglobin level <12g/dL, absolute neutrophil count <1.50×109/L, and platelet count (PLT) <100×109/L. Pancytopenia was considered when the three hematopoietic lineages were affected. Haptoglobin values were not used in this study.
Statistical analysisOverall survival (OS) was calculated from the date of diagnosis to the date of death or the date of last follow-up. Survival analysis was performed using the Kaplan–Meier method. The distributions of the presentation of characteristics were compared between the three subcategories and between Classic PNH and PNH-sc/AA by the chi-squared or Fisher's exact test as necessary for categorical variables, and by the Kruskal–Wallis (three subcategories) or Mann–Whitney (two groups) test for continuous variables. The percentages of GPI-AP deficient red blood cells and granulocytes were compared using Student's t-test. All statistical analyses were performed using the Statistica v.8 program. A p-value <0.05 was considered statistically significant.
Ethical approvalThe Ethics Committee of the Hospital de Clinicas, Universidade Federal do Paraná, Brazil approved the study.
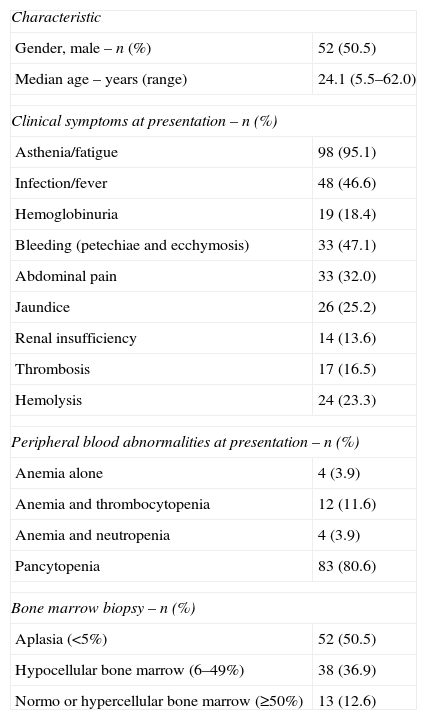
ResultsPatient characteristicsOf the 398 patients tested, 125 (31.4%) had a detectable PNH population. Of these, 103 (51 male and 52 female), who had clinical data available, were studied. The median age at presentation was 24.1 years (range: 5.5–62.0 years). There were 24 patients below the age of 18 years (range: 5.5–17.9 years; median: 14.7). All these young patients were in the aplastic groups (11 HPNsc/AA and 13 HPN/AA).
Hemoglobinuria was identified during monitoring in 43 (41.7%) patients, while it was the initial symptomatic manifestation in only 19 (18.4%) patients. The frequencies of infections and bleeding were 46.6% and 47.1%, respectively. Ninety-eight patients (95.1%) presented with asthenia, 33 (32.0%) with abdominal pain, and 14 (13.6%) with renal insufficiency; 17 (16.5%) developed thrombosis during monitoring and 49 patients (47.6%) had documented hemolysis. Twenty-six patients (25.2%) with aplasia at diagnosis developed the PNH clone and hemolysis in a median of 2.35 years after diagnosis. Of these patients, eleven (10.7%) developed hemolysis five years after the diagnosis of aplasia.
Peripheral blood abnormalities were present in 101 patients: 83 (80.6%) had pancytopenia, 12 (11.6%) anemia and thrombocytopenia, and four (3.9%) had anemia and leukopenia. The median hemoglobin level was 8.8g/dL (range: 3.8–14.5g/dL), the median absolute neutrophil count was 0.94×109/L (range: 0.26–1.45×109/L) and the median platelet count was 25×109/L (range: 2–294×109/L). The median LDH concentration was 328U/L (range: 30–7970U/L; normal range 190–240U/L). Ninety patients (87.4%) had hypocellular bone marrow.
Patients included in this study were divided into the subcategories of Classic PNH (10 patients), PNH/AA (39 patients), and PNH-sc/AA (54 patients) based on the proposed PNH working clinical classification.1 The initial characteristics of the subgroups are summarized in Table 1.
Patient characteristics (n=103).
| Characteristic | |
| Gender, male – n (%) | 52 (50.5) |
| Median age – years (range) | 24.1 (5.5–62.0) |
| Clinical symptoms at presentation – n (%) | |
| Asthenia/fatigue | 98 (95.1) |
| Infection/fever | 48 (46.6) |
| Hemoglobinuria | 19 (18.4) |
| Bleeding (petechiae and ecchymosis) | 33 (47.1) |
| Abdominal pain | 33 (32.0) |
| Jaundice | 26 (25.2) |
| Renal insufficiency | 14 (13.6) |
| Thrombosis | 17 (16.5) |
| Hemolysis | 24 (23.3) |
| Peripheral blood abnormalities at presentation – n (%) | |
| Anemia alone | 4 (3.9) |
| Anemia and thrombocytopenia | 12 (11.6) |
| Anemia and neutropenia | 4 (3.9) |
| Pancytopenia | 83 (80.6) |
| Bone marrow biopsy – n (%) | |
| Aplasia (<5%) | 52 (50.5) |
| Hypocellular bone marrow (6–49%) | 38 (36.9) |
| Normo or hypercellular bone marrow (≥50%) | 13 (12.6) |
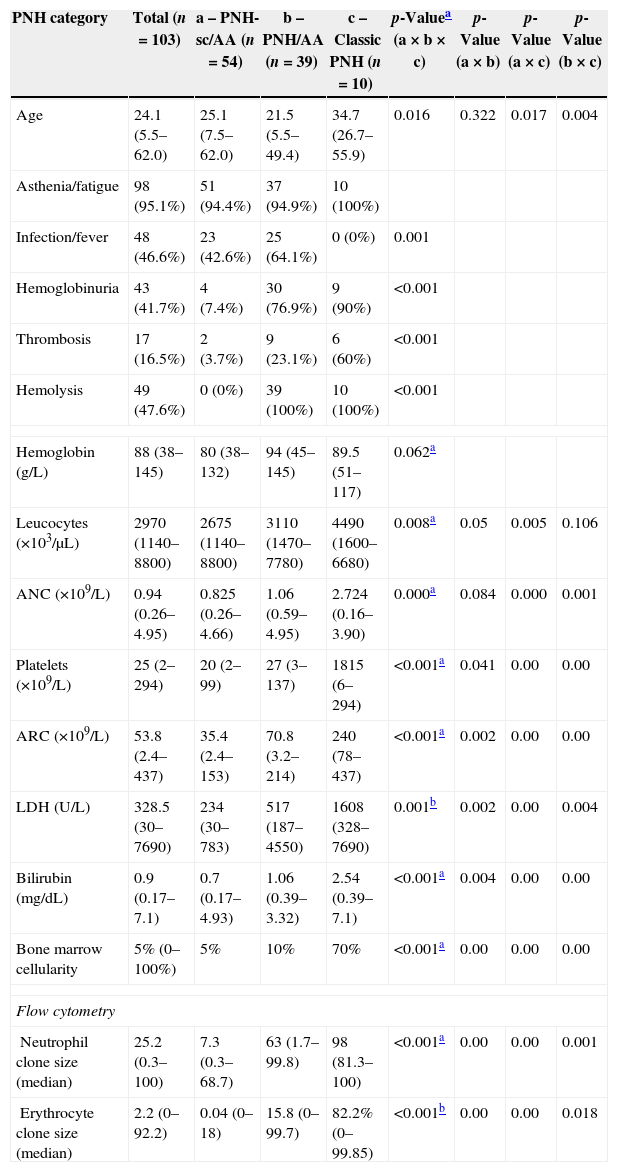
The median ages of PNH-sc/AA, PNH/AA and Classic PNH patients were 25.1, 21.5 and 34.7 years, respectively (p-value=0.016). The clinical symptoms of hemolysis, hemoglobinuria and thrombosis were higher in the hemolytic forms of PNH (PNH/AA and Classic PNH) than in PNH-sc/AA (p-value<0.001), whereas infection and fever were higher in the PNH-sc/AA group (p-value<0.001). The median numbers of neutrophils and platelets were significantly lower in the aplastic groups than in the Classic PNH group (p-value for both <0.001). On the other hand, the Classic PNH group had higher hemolytic markers, such as LDH (p-value=0.001), reticulocyte count (p-value <0.001) and bilirubin levels (p-value <0.001 – Table 2).
PNH subgroup characteristics.
| PNH category | Total (n=103) | a – PNH-sc/AA (n=54) | b – PNH/AA (n=39) | c – Classic PNH (n=10) | p-Valuea (a×b×c) | p-Value (a×b) | p-Value (a×c) | p-Value (b×c) |
|---|---|---|---|---|---|---|---|---|
| Age | 24.1 (5.5–62.0) | 25.1 (7.5–62.0) | 21.5 (5.5–49.4) | 34.7 (26.7–55.9) | 0.016 | 0.322 | 0.017 | 0.004 |
| Asthenia/fatigue | 98 (95.1%) | 51 (94.4%) | 37 (94.9%) | 10 (100%) | ||||
| Infection/fever | 48 (46.6%) | 23 (42.6%) | 25 (64.1%) | 0 (0%) | 0.001 | |||
| Hemoglobinuria | 43 (41.7%) | 4 (7.4%) | 30 (76.9%) | 9 (90%) | <0.001 | |||
| Thrombosis | 17 (16.5%) | 2 (3.7%) | 9 (23.1%) | 6 (60%) | <0.001 | |||
| Hemolysis | 49 (47.6%) | 0 (0%) | 39 (100%) | 10 (100%) | <0.001 | |||
| Hemoglobin (g/L) | 88 (38–145) | 80 (38–132) | 94 (45–145) | 89.5 (51–117) | 0.062a | |||
| Leucocytes (×103/μL) | 2970 (1140–8800) | 2675 (1140–8800) | 3110 (1470–7780) | 4490 (1600–6680) | 0.008a | 0.05 | 0.005 | 0.106 |
| ANC (×109/L) | 0.94 (0.26–4.95) | 0.825 (0.26–4.66) | 1.06 (0.59–4.95) | 2.724 (0.16–3.90) | 0.000a | 0.084 | 0.000 | 0.001 |
| Platelets (×109/L) | 25 (2–294) | 20 (2–99) | 27 (3–137) | 1815 (6–294) | <0.001a | 0.041 | 0.00 | 0.00 |
| ARC (×109/L) | 53.8 (2.4–437) | 35.4 (2.4–153) | 70.8 (3.2–214) | 240 (78–437) | <0.001a | 0.002 | 0.00 | 0.00 |
| LDH (U/L) | 328.5 (30–7690) | 234 (30–783) | 517 (187–4550) | 1608 (328–7690) | 0.001b | 0.002 | 0.00 | 0.004 |
| Bilirubin (mg/dL) | 0.9 (0.17–7.1) | 0.7 (0.17–4.93) | 1.06 (0.39–3.32) | 2.54 (0.39–7.1) | <0.001a | 0.004 | 0.00 | 0.00 |
| Bone marrow cellularity | 5% (0–100%) | 5% | 10% | 70% | <0.001a | 0.00 | 0.00 | 0.00 |
| Flow cytometry | ||||||||
| Neutrophil clone size (median) | 25.2 (0.3–100) | 7.3 (0.3–68.7) | 63 (1.7–99.8) | 98 (81.3–100) | <0.001a | 0.00 | 0.00 | 0.001 |
| Erythrocyte clone size (median) | 2.2 (0–92.2) | 0.04 (0–18) | 15.8 (0–99.7) | 82.2% (0–99.85) | <0.001b | 0.00 | 0.00 | 0.018 |
a: PNH-sc/AA group; b: PNH/AA group; c: Classic PNH group.
ANC: absolute neutrophil count; ARC: absolute reticulocyte count; LDH: lactate dehydrogenase.
FCM data showed that larger PNH clones were associated with classical PNH and thrombotic events, while smaller PNH clones were associated with bone marrow aplasia. At diagnosis PNH clone sizes in red cells, granulocytes and monocytes were significantly different between the three clinical subcategories (p-value <0.001) for all the studied monoclonal antibodies (CD55, CD59, CD14, CD16, CD24 and CD66b). The median PNH clone percentages in red cells were 0.04% (range: 0–18.0%), 15.8% (range: 0–99.7%) and 82.2% (range: 0–99.85%) in PNH-sc/AA, PNH/AA and Classic PNH, respectively (p-value <0.001). Moreover, the median PNH clone percentages in granulocytes were 7.3% (range: 0.3–68.7%), 63.0% (range: 1.7–99.8%) and 98.0% (range: 81.3–100.0%) in PNH-sc/AA, PNH/AA and Classic PNH, respectively (p-value <0.001 – Figures 1–3).


Number of patients with predominantly small erythrocyte clones in paroxysmal nocturnal hemoglobinuria with subclinical features and aplastic anemia, intermediate clones in paroxysmal nocturnal hemoglobinuria with aplastic anemia and large clones in classic paroxysmal nocturnal hemoglobinuria.

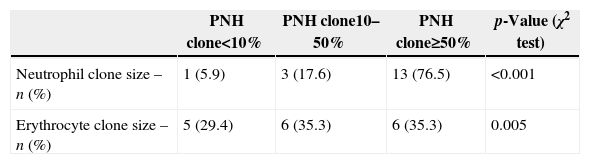
Seventeen patients presented with thrombotic events: six in the Classic PNH group, nine in the PNH/AA group and two in the PNH-sc/AA group. There were five cases of deep vein thrombosis (DVT), five abdominal thromboses, five arterial thromboses with ischemic stroke, one sinus venous thrombosis and one renal vein thrombosis. Two PNHsc/AA patients had DVT despite the absence of hemolytic symptoms, and died. Four of the PNH/AA patients died due to thrombotic events. Regarding the PNH clone, clones larger than 50% were seen in neutrophils of 13 (76.5%) patients (p-value <0.001), and in erythrocytes of six (35.3%) patients (p-value=0.005 – Table 3).
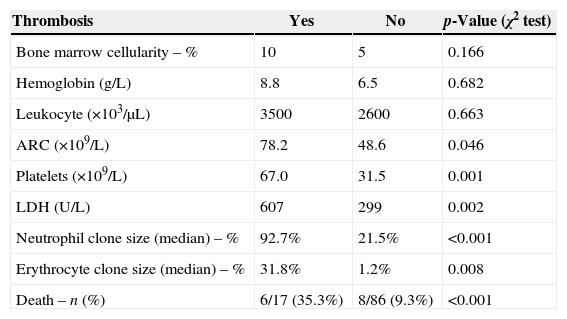
The differences between patients with or without thrombotic events are shown in Table 4. There were statistically significant differences in the number of platelets (p-value=0.001), LDH (p-value=0.002) and in median percentages of the PNH clone in neutrophils (p-value <0.001) and erythrocytes (p-value=0.008). The median neutrophil PNH clone in thrombotic patients was 92.7% (range: 3.4–100%) vs. 21.8% (range: 0.25–99.9%) in non-thrombotic patients. The median erythrocyte PNH clone was 31.8% (range: 0.0–97.1%) vs. 1.2% (range: 0.0–92.2%) in thrombotic and non-thrombotic patients, respectively. The bone marrow cellularity, hemoglobin concentration, leukocyte count and absolute reticulocyte count were similar between the two groups. There were six deaths among patients with thrombotic events (35.3%), and eight (9.3%) among the non-thrombotic group.
Characteristics of patients with and without thrombosis.
| Thrombosis | Yes | No | p-Value (χ2 test) |
|---|---|---|---|
| Bone marrow cellularity – % | 10 | 5 | 0.166 |
| Hemoglobin (g/L) | 8.8 | 6.5 | 0.682 |
| Leukocyte (×103/μL) | 3500 | 2600 | 0.663 |
| ARC (×109/L) | 78.2 | 48.6 | 0.046 |
| Platelets (×109/L) | 67.0 | 31.5 | 0.001 |
| LDH (U/L) | 607 | 299 | 0.002 |
| Neutrophil clone size (median) – % | 92.7% | 21.5% | <0.001 |
| Erythrocyte clone size (median) – % | 31.8% | 1.2% | 0.008 |
| Death – n (%) | 6/17 (35.3%) | 8/86 (9.3%) | <0.001 |
LDH: lactate dehydrogenase; ARC: absolute reticulocyte count.
As a retrospective study, the cohort of patients received different treatments depending on the physician and time criteria. The majority of patients (81.9%) of aplastic groups (PNH-sc/AA and PNH/AA) received cyclosporine A-based immunosuppressive therapy with or without corticosteroids some time during the follow-up, and 12 patients (12.7%) received anti-human thymocyte immunoglobulin immunosuppressive therapy. Hematopoietic stem cell transplantation (HSCT) was performed in 27 patients (28.7%), the majority of whom were in the PNH-sc/AA group. Nine PNH/AA and two Classic PNH patients received HSCT due to hemolytic symptoms. Eculizumab was introduced in 2010, and five patients received this drug until the conclusion of the study.
The median follow-up period for all patients was 49.2 months (range: 2.4–310 months), with an OS of 81.7% at ten years after diagnosis. There was a slight difference in survival between PNH-sc/AA and the other two groups (76.5% vs. 87.9%; p-value=0.112). In total 14 patients died, nine in the PNH-sc/AA group and five in the PNH/AA group. The causes of death were sepsis (six patients), pulmonary thromboembolism (four patients), mesenteric thrombosis (two patients), severe graft-versus-host disease (GVHD) after HSCT (one patient) and sudden death (one patient). No death was reported in the classical PNH group by the end of the follow-up period.
DiscussionPNH is a rare acquired disorder of hematopoietic stem cells, which is characterized by a highly variable clinical course, including intravascular hemolysis, bone marrow failure syndromes and thrombosis.1–3 The natural history of PNH has been widely discussed in the literature in retrospective series,3,4,8,9,16,19,20 which confirm the association between PNH and bone marrow syndromes. The proportion of patients with bone marrow hypoplasia who develop clinical or subclinical PNH varies from 22%8 to 40%.16 Some studies that used Ham's test to diagnose PNH showed that 5–10% of patients with Classic PNH develop pancytopenia.2,4
The current study identified 103 patients with a PNH clone, 87.4% of whom had hypoplastic marrow at diagnosis. PNH-associated cytopenias were common and recurrent infections and bleeding were seen in 46.6% and 47.1% of patients, respectively. The high prevalence of bone marrow failure may be a result of an ascertainment bias explained by the fact that the Hospital de Clinicas in Curitiba receives cases of aplasia from many regions of Brazil. For the same reason the majority of patients (81.9%) received immunosuppressive therapy and 28.7% received HSCT.
Patients included in this study were assigned to three subcategories based on the proposed PNH working clinical classification,1 namely Classic PNH, PNH/AA, and PNH-sc/AA. As expected, the clinical symptoms of hemolysis, hemoglobinuria and thrombosis were higher in the hemolytic forms of PNH (PNH/AA and Classic PNH) than in PNH-sc/AA, whereas infection and fever were higher in the latter group. There were significant differences in terms of peripheral blood cell counts between the three subcategories, especially between the Classic PNH and PNH-sc/AA subcategories. The median PNH clone size was also significantly different between the three clinical subcategories at diagnosis. On the other hand, eight patients in the PNH/AA group presented with intense hemolytic symptoms and PNH clones greater than 50%, despite the hypocellular bone marrow, suggesting an overlap between the subgroups. This fact could be explained by the heterogeneity of the sample and the disease, and by the retrospective bias of this study.
PNH mainly presents as a disease of adults, but the median age found in the current series was lower than in the literature (24.1 vs. 34.2 years).2,3,19 This difference can be explained by the proportion of aplastic patients who evolved with the PNH clone (PNH-sc/AA) after a long period of time in our series. However, if the median age of the Classic PNH group is considered, it is similar to other studies.4,16,20
In the last decade, the detection of GPI-AP deficient cells by flow cytometry greatly increased the sensitivity of detecting a PNH clone in both red blood cells and leukocytes, minimizing the effects of red cell transfusions on estimating clone size.5,11 Since the introduction of FCM, quantitative and kinetic differences of GPI-AP deficient clones have been reported between PNH patients with and without marrow failure. The literature suggests that the clinical manifestations due to chronic hemolysis in PNH appear to be more common in patients with large populations of cells deficient in GPI proteins.1,19–21 Pu et al. showed a direct relationship between the size of PNH clone and the development of intravascular hemolysis in patients with aplasia.19 In the current study large granulocyte PNH clones were found in Classic PNH (98.0%), medium clones in PNH-AA (63.0%) and small PNH clones in PNH-sc/AA (7.3%), suggesting that larger PNH clones (>50%) are associated with increased risks of hemoglobinuria and thrombosis, whereas small PNH clones (<50%) are associated with bone marrow failure.
In this study, life-threatening thrombotic complications were present in 16.5% of patients, with six deaths occurring in this group (35.3% vs. 9.3% in the non-thrombotic group). All three subcategories had at least one episode of thrombosis. Two PNHsc/AA patients had DVT despite the absence of hemolytic symptoms, and died. Four of the PNH/AA patients died due to thrombotic events. PNH clones larger than 50% were seen in the granulocytes of 13 (76.5%) thrombotic patients, which is consistent with the literature that says that a clone size greater than 50% is associated with increased risk of thrombosis and the need of primary anticoagulation.7,21 In the current cohort the size of the PNH clone was greater in patients with thrombosis for both neutrophils (92.7% vs. 21.5%) and erythrocytes (31.8% vs. 1.2%).
The OS ten years after diagnosis was 81.7% in this study, which is slightly higher than other studies,20 which reported 77.6% at 10 years. Infection and thrombotic events were identified as the main causes of death. There were 14 deaths (13.5%), 12 of which were attributable to PNH or aplasia; of the deaths, six (42.8%) were secondary to thrombosis. With regard to the disease subcategories, the long-term outcomes were similar between patients in the hemolytic PNH subcategories, but slightly different in PNH-sc/AA group, probably due to a high risk of infection. Further studies may shed insights into the hypothesis that the PNH-sc/AA subcategory might be a separate disease entity from Classic PNH.1,5,22
ConclusionThis retrospective study of 103 PNH patients over an 11-year period represents the largest collection of such patients from a single center in Brazil. The results confirmed the suggestion that PNH is not a simple binary diagnosis and both flow cytometric characterization of GPI-AP expression on peripheral blood cells and marrow analysis are required for comprehensive disease classification. FCM data from this study shows that larger PNH clones are associated with classical PNH symptoms and increased risk of thrombosis, even in patients with bone marrow failure, whereas smaller PNH clones are associated with bone marrow aplasia.
Conflicts of interestThe authors declare no conflicts of interest.
The authors wish to thank Rosana Inara Cattaneo, Miriam Perlingeiro Beltrame, Noely Silva and Leila Oliveira for technical assistance with flow cytometric studies. Thanks to Dr. Adam Campbell Smith for the English revision and to Dr. Alberto Orfao for the scientific comments.

