


About 10% of sickle cell anemia patients will have ischemic stroke. Adams showed stroke incidence reduction in children receiving monthly erythrocyte transfusions by reducing transcranial Doppler (TCD) velocities. Since then, chronic transfusion is recommended as primary stroke prophylaxis. This study aims to assess the effectiveness of chronic transfusions as stroke prophylaxis.
MethodRetrospective study, reviewing medical records from 15 sickle cell anemia patients undergoing chronic transfusion. Data collected were age, sex, adverse reactions, stroke, hemoglobin, reticulocytes, ferritin, HbS and TCD values (baseline, after 12 and 24months of treatment).
ResultsThe mean age was 118.67±41.40 months; six patients experienced allergic reactions. No stroke was recorded. One patient had alloimmunization. There was a decrease in the HbS rate and an increase in hemoglobin values in the first 12months. Values were maintained after 24months, but with no improvement of data. Before treatment, the mean HbS rate was 75.18%±11.69; after 12months, 41.63±14.99 and after 24months, 43.78±10.6. Thirteen patients initiated chelation after 12months from the beginning of chronic transfusions and ferritin decline after 24months. Pre-transfusional TCD velocities were 204.28±9.41cm/s (right) and 198.85±33.37cm/s (left). After a 12-month treatment, these values were 158.5±28.89cm/s and 157.62±34.43cm/s, respectively, and this reduction was statistically significant (p=0.002 right and p=0.02 left). After 24months, these values were 149.63±26.95cm/s (right) and 143.7±32.27cm/s (left).
ConclusionSignificant reduction of TCD velocity occurred after treatment with chronic transfusion in sickle cell anemia patients, leading to a normal or conditional test and reducing stroke risk in all but one patient.
Sickle cell disease (SCD) is the most common severe monogenetic disorder in the world. It is estimated that 25–30 thousand people in Brazil live with this diagnosis. The new case incidence is approximately 3500 cases a year.1 The presence of hemoglobin S, in situations of hypoxia or dehydration can lead to its polymerization, causing erythrocyte rigidity and vascular occlusion, which is the central pathophysiology of the disease. Chronic hemolysis is a hallmark of the disease and is both intra- and extravascular.2
Stroke physiopathology in SCD implicates in sickled red blood cell, causing acute vascular occlusion, associated with previous vasculopathy, mainly in distal intracranial internal carotid artery segments and proximal portions of the middle and anterior cerebral arteries. Vasculopathy results from fibroblast proliferation in the vascular intima. This narrowing appears in MRI as much as in transcranial Doppler ultrasonography (TCD) due to rising blood flow velocities in the studied vessels.3 Thus, since it is less expensive, offers less risk to patients and is highly accurate, the TCD is widely used as stroke risk assessment.3,4
One in 10 sickle cell disease patients will suffer a stroke,5 this being one major cause of death in this population.6 The isquemic form is predominant in children, whereas the hemorrhagic form is more common in adulthood. There’s a bimodal peak incidence between 2–5 years and after 29 years, with decreased incidence from 10 to 19 years.7,8 Most pediatric patients recover without physical sequelae after proper treatment, nevertheless cognitive sequelae may persist.8 Therefore, focus on primary prevention is important.
In 1998, the STOP study found a 92% reduction in stroke incidence in children undergoing chronic transfusion. Higher risk patients were found by TCD values; they must not have had a previous cerebrovascular event. Since its publication, chronic transfusion is recommended as primary stroke prophylaxis.9 These patients also had fewer vaoscular occlusive crises (VOC) and acute chest syndrome (ACS) episodes.5 Another retrospective study showed that TCD screening and use of regular transfusions in high-risk patients decreased the annual stroke rate from 0.44 to 0.19 per 100 person-years.10 Still, risks associated with this therapy, such as alloimmunization, iron overload and disease transmission, are relevant.
This study aims to assess chronic transfusion effectiveness as primary stroke prophylaxis in sickle cell disease patients.
MethodThis is a retrospective study, approved by the Ethics Committee of the Universidade Federal de São Paulo – UNIFESP (CAAE: 68549617.3.0000.5505). Medical records from 23 sickle cell disease patients undergoing chronic transfusion, from January 2008 until June 2016 were reviewed. Laboratorial tests data were found in electronic charts. Patients with less than 12months of treatment, incomplete data or previous stroke were excluded.
Sickle cell disease patients (HbSS and HbSβ0) between 2–16 years are screened annually for stroke risk with TCD, as per global consensus, by the Neurovascular Unit of the hospital. Patients with 2 abnormal tests are referred to chronic transfusion therapy; the Pediatric Hematology unit offers comprehensive SCD care, including chronic blood transfusion. In our hospital, we use simple red blood cell transfusion, aiming to reduce the HbS rate below 50% and to keep the pre-transfusional mean baseline hemoglobin below 10g/dL to avoid hyperviscosity. Patients with overt stroke were referred to chronic transfusion as well for secondary stroke prophylaxis. The TCD was performed in accordance with the Stroke Prevention Trial in Sickle Cell Anemia (STOP) Protocol.9 Chelation was initiated after 3 ferritin level measured above 1000ng/mL.
The data collected were age, sex, date of the beginning of treatment and its clinical indication, presence or absence of transfusion-related reactions (such as alloimmunization, allergic reaction, hemolytic reaction), presence or absence of stroke, HbS, hemoglobin and serum ferritin values at the beginning of treatment and after 12 and 24month of chronic transfusions and TCD values before and after 12 and 24months of treatment.
A new neurologic event was considered compatible after evaluation by a pediatric neurologist and suggestive radiological imaging.8 Acute chest syndrome was defined as the presence of respiratory symptoms associated with new radiological imaging.11
Statistical analysis was made with descriptive variables, including means and standard deviations (SD) to describe the patients’ characteristics. The Student's t test was used to compare means between the TCD velocities, and hemoglobin and sickle hemoglobin before and after chronic transfusion. The serum ferritin level did not present normal distribution. Therefore, statistical evaluation was made with the Wilcoxon Ranked Test. The level of significance was set at p < 0.05. The STATA/SE version 11.2 was used for analysis.
ResultsFrom the 23 selected patients, 8 were excluded because of previous stroke. Fifteen patients were included (HbSS). Their mean age was 118.67±41.40 months; 40% were male and 60% were female. The mean transfusion treatment period was 45.60±11.28 months. Six patients experienced adverse reactions (40%); all of them were allergic reactions. No stroke was recorded. Alloimmunization was observed in only one patient (6.67%).
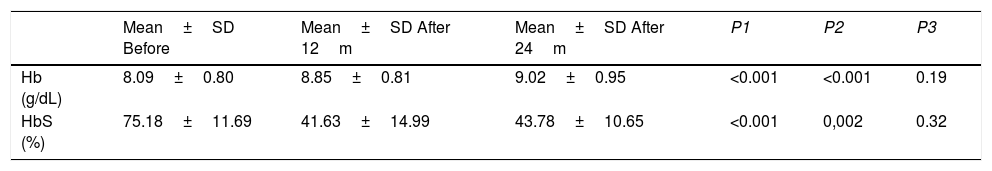
The sickle hemoglobin percentage and hemoglobin baseline, before and after 12 and 24months of chronic transfusion, are shown in Table 1. There was a significant decrease in the HbS rate and a significant increase in hemoglobin in the first 12months. The values were maintained after 24months, but with no improvement of data.
Hemoglobin and sickle hemoglobin before and after chronic transfusion.
| Mean±SD Before | Mean±SD After 12m | Mean±SD After 24m | P1 | P2 | P3 | |
|---|---|---|---|---|---|---|
| Hb (g/dL) | 8.09±0.80 | 8.85±0.81 | 9.02±0.95 | <0.001 | <0.001 | 0.19 |
| HbS (%) | 75.18±11.69 | 41.63±14.99 | 43.78±10.65 | <0.001 | 0,002 | 0.32 |
Student’s t test.
P1: before x after 12months of transfusions.
P2: before x after 24months of transfusions.
P1: after 12months of transfusions x after 24months of transfusions.
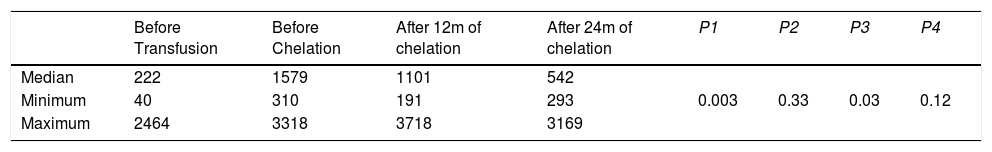
Only 2 patients have not experienced iron overload. Thirteen left initiate chelation with deferasirox after approximately 12months from the beginning of chronic transfusions (range 6–40 months). Chelation did not show a significant effect on ferritin rates in the first 12months after treatment. However, it has a statistically significant effect on the ferritin decline after at least 24months and it can be compared to levels before transfusion. Data is shown on Table 2 and in Fig. 1.
Serum ferritin values before transfusion, before chelation and after 12 and 24months of chelation.
| Before Transfusion | Before Chelation | After 12m of chelation | After 24m of chelation | P1 | P2 | P3 | P4 | |
|---|---|---|---|---|---|---|---|---|
| Median | 222 | 1579 | 1101 | 542 | 0.003 | 0.33 | 0.03 | 0.12 |
| Minimum | 40 | 310 | 191 | 293 | ||||
| Maximum | 2464 | 3318 | 3718 | 3169 |
Wilcoxon Ranked Test.
P1 = before transfusion x before chelation.
P2 = before chelation x after 12months of chelation.
P3 = before chelation x after 24months of chelation.
P4 = before transfusion x after 24months of chelation.

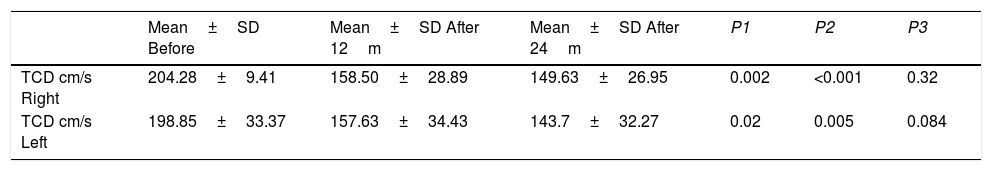
TCD flow velocities rates before and after treatment are in Table 3. After 12months of treatment, there was a significant reduction in velocities. After 24months of transfusions, these values were still dropping. However, when data between 12 and 24months were compared, there was no significant velocity reduction. After at least 24months of chronic transfusions, three patients persisted with conditional TCD, one patient maintained abnormal velocities and all others changed to normal.
Transcranial Doppler ultrasonography (TCD) velocities before and after chronic transfusion.
| Mean±SD Before | Mean±SD After 12m | Mean±SD After 24m | P1 | P2 | P3 | |
|---|---|---|---|---|---|---|
| TCD cm/s Right | 204.28±9.41 | 158.50±28.89 | 149.63±26.95 | 0.002 | <0.001 | 0.32 |
| TCD cm/s Left | 198.85±33.37 | 157.63±34.43 | 143.7±32.27 | 0.02 | 0.005 | 0.084 |
Student’s t test.
P1: before x after 12months of transfusions.
P2: before x after 24months of transfusions.
P1: after 12months of transfusions x after 24months of transfusions.
Adams et al. saw a 92% reduction in stroke risk for chronic transfused patients in their STOP study.9 In 2016, the TWiTCH study was published. It tested the hydroxycarbamide (hydroxyurea) non-inferiority after chronic transfusion therapy withdrawal in maintenance of normal TCD velocities in primary sickle cell disease prophylaxis. Patients were followed for 24months after randomization, with periodic TCD tests. In both arms, TCD velocities were kept normal. The alternative arm achieved iron overload reduction, while it remained the same in the standard group. No stroke was reported. Therefore, the authors concluded that hydroxyurea is an alternative for primary stroke in sickle cell disease prophylaxis for patients with at least 1year of chronic transfusion and no vasculopathy.12
In this study, there was important decrease in TCD velocities after chronic transfusion treatment in sickle cell disease patients. It turned an abnormal into a normal or conditional test in all but one patient, suggesting lower stroke risk. However, TDC rates have not continued dropping, but rather have only been maintained.
The advantages of maintaining chronic transfusion therapy are undeniable and very well established in the current literature. However, the viability of maintaining this practice in most Brazilian health centers is questionable, since a minimum technological arsenal is needed, and this is not yet available throughout the country.
Aygun et al. made an important observation regarding chronic transfusion therapy: they claimed that in a real world situation it is difficult to maintain HbS <30% and that maybe this would not necessarily do harm to patients, since values above 30% were verified while conducting the TWiTCH study and even in the STOP study. It was found that the mean pre-transfusion HbS of chronic transfusion patients in the most important academic health centers in the United States was 34%, while the 75th and 90th percentiles were 41% and 50%, respectively. It was also found that the most important variables to achieve HbS <30% were low age, less time on chronic transfusion and whether the transfusions had occurred on the correct dates (4 weeks after the last transfusion). The study highlighted technical difficulties in following the guidelines given by large studies, taking into account obstacles such as adherence of the patient and his or her family in attending to the correct transfusion date, hypersplenism, development of auto- and alloantibodies, and obtainment of peripheral venous access.13
In this study, patients haven’t achieved HbS below 30% after chronic transfusions. Apheresis machines were not available in our hospital and we do not have enough staff members to perform exchange transfusion. Due to this, only simple transfusion is offered to all patients. Only one patient had an alloantibody and the hypersplenism was not documented. It is presumed that an HbS rate over 30% may be due to an incomplete marrow suppression, maintaining the HbS production.
Another important criticism of chronic transfusion therapy is its adverse effects, mainly such as iron overload and alloimmunization. Ethnic disparities between blood recipients and donors increase the risk of developing alloantibodies. It is known that the antibodies produced are generally against antigens prevalent in the white population, the predominant donor ethnicity, whereas the receptors are mostly black. In Brazil, the profile of blood donors in 2014 was found to be 49.45% white, 37.48% brown, and only 11.87% black.14 In Uganda and Jamaica, where the population is more uniform, alloimmunization rates are lower: 6.1% and 2.6%, respectively. In the USA, sickle-cell alloimmunization rates range from 20 to 50%, but among thalassemic patients (who have greater ethnic compatibility with blood donors) this rate is 10%. The earlier initiation of transfusions, as in the case of thalassemia, may possibly induce greater antigenic tolerance in the blood receptors.15
A study performed in the state of Alagoas, published in 2011, showed that the prevalence of alloimmunization in patients with the SS genotype was 12.7%. Seventy percent of the antibodies belonged to the Rh and Kell blood groups. The median age was 11.5 years.16 In Minas Gerais, in 2005, a group saw 9.9% alloimmunization in patients with sickle cell disease with 23.3 years as median age, 79% of which belonged to the Rhesus and Kell systems.17 Another study conducted in São Paulo, but with a median age of 25 years, found an alloimmunization rate among sickle cell disease patients of 22.6%. The anti-Kell antibody was the most frequent (7.5%), followed by the anti-C (5.7%).18 These data reinforce the need to use phenotyped components in order to reduce alloimmunization rates. Vichinsky et al. found a reduction in the alloimmunization ratio using extended compatibility for the antigens of the Rh (D, C, E) and Kell groups.19
Chou et al., in a study published in 2013, found that red blood cell transfusions from African-descent donors failed to reduce the alloimmunization rate. In this study, a high rate of unexplained Rh antibodies was verified, for example, in patients positive for an antibody-specific antigen or antibody formation in patients negative for a given antigen and who had received negative blood transfusion for the same antigen. Thirty-five percent of these unexplained antibodies had a clinical repercussion, with a delayed transfusion reaction. These clinically significant antibodies occurred not only in multi-transfused patients, but also in patients transfused only one time, suggesting that chronic transfusion patients remain at risk of developing significant alloantibodies, contrary to previous reports that found such antibodies only up to 6 months after the chronic transfusion had started. High-resolution genotyping of these patients revealed that 87% inherited at least one Rh variant allele, which could potentially encode altered or partial antigens that may not be distinguished from traditional antigens by simple serological tests.20,21
Thus, it is suggested that the best practice is prior patient genotyping and transfusion of compatible blood components. However, due to the high cost, this practice is not yet widely available in Brazilian public health services. In this study, extended phenotyping was done for all patients before the first transfusion and alloimmunization was seen in only one patient (6.67%). Nevertheless, not using genotyping for a completely compatible transfusion did not elevate the alloimmunization ratio in this study. Other factors may have contributed to lower the alloimmunization rate, as the transfusions were always performed at the same institution, avoiding the risk of transfusions done at institutions not providing extended matching. Furthermore, children with SCD who are chronically transfused might have less chronic inflammation and may have a decreased immunologic response to alloantigens.17
Another major disadvantage of chronic transfusion is the imminent iron overload. However, with the use of oral iron chelators and the possibility of transition to therapy with hydroxyurea after 1year of blood transfusion, this adversity can be overcome.
Hepatic iron overload can lead to fibrosis and inflammation, leading to elevated risk of cirrhosis and hepatocarcinoma. Therefore, iron chelation in polytransfused patients is mandatory. Patients should be screened for serum ferritin levels and must initiate chelation when levels are above 1000μg/L. Ferritin, however, is not a good marker for chelation response; a prospective study found a significant decrease in liver iron concentration (LIC) after 1year of chelation, whereas a decrease in ferritin values was only achieved after 5 years. In spite of this, in the Brazilian public health system we do not have another way to manage iron overload other than ferritin levels. The use of oral iron chelation (deferasirox) provides good patient compliance.15,22
Even today, chronic transfusion is the only proven therapy capable of preventing stroke in sickle cell children. Recent studies have demonstrated the efficacy of hydroxyurea in maintaining TCD velocities stable, but patients at higher risk (with abnormal TCD velocity) should have at least 1year of chronic blood transfusion. These studies, although promising, were short-term studies (24months), and, therefore, longer ones are needed to confirm these benefits. Since we are dealing with adverse effects as deleterious as those resulting from chronic transfusion, we are forced to seek new alternatives to this therapy.
The major limitations of this study were its small sample size and retrospective nature.
ConclusionRegardless of its small sample size and retrospectively collected data, this study showed that chronic transfusion was as effective as primary stroke prevention in high-risk sickle cell disease patients. Significant reduction of the TCD velocity occurred after treatment with chronic transfusion in sickle cell disease patients, leading to a normal or conditional test and reducing the stroke risk for the majority of patients.
Conflicts of interestThe authors declare no conflicts of interest.
The authors would like to thank the Neurovascular Unit in the Department of Neurology and Neurosurgery, Universidade Federal de São Paulo, São Paulo, Brazil, for performing transcranial Doppler ultrasonography on our patients. We also would like to thank Doctor Iberê Pereira Datti who was incredibly helpful with data analysis.


