


Hemoglobin (Hb) is the main protein in red blood cells, composed by a tetrameric structure with 2 α and 2 β chains. Dysfunctions in this molecule result in a group of diseases called hemoglobinopathies comprising Hb variants and thalassemia. More than 1000 hemoglobin variants have been identified so far, affecting approximately 7 % of the world population.1 Common and rare Hb variants were found in the Brazilian population, but usually with no clinical repercussions. Hb Maputo is caused by the point mutation c.142G>T, leading to the amino acid exchange p.Asp47Tyr of the β chain. It was first described in 1983 in a patient from Maputo–Mozambique with a severe anemia condition associated with HbS.2 Later, in 1991, a US family was identified carrying the heterozygous mutation with no clinical nor laboratory impact.3 In Brazil, Hb Maputo was reported in 2013 in 2 newborn individuals and their mothers from Minas Gerais, all of them in heterozygosity and with no clinical and laboratory consequences.4 The HBB:c.9T>C variant is a silent mutation, commonly found in the healthy population, without any pathological relationship.5,6 This Hb variant does not affect glycated hemoglobin A1c (HbA1c) and does not impair Diabetes mellitus diagnosis and monitoring.7 This work aimed to describe an in-depth study with hematological, biochemical, and molecular analysis of a family from Duque de Caxias, the nearby city of Rio de Janeiro-Brazil, bearing Hb Maputo and HBB:c.9T>C variant.
MethodsBlood samples from four individuals of the same family, a 3 m/o male, 35 y/o father, 31 y/o mother, and 27 y/o paternal aunt, were collected by vacuum venipuncture and used for biochemical laboratory tests performed by Labmax Plenno (Labteste, Buenos Aires, Argentina), Hb-HPLC (Variant I Express, Bio-Rad, Japan), vitamin B12, folic acid, alkaline Hb electrophoresis and osmotic fragility curve (OFC) with and without incubation at 37 °C for 24 h by spectrophotometry. Molecular analyzes were performed for the detection of α thalassemia (deletions α3.7 kb, α4.2 kb and α-SEA) by multiplex polymerase chain reaction (PCR) (T100 Thermal Cycler, BioRad, Singapora) and by automated Sanger DNA sequencing for detection of β thalassemia-associated point mutations IVS1–1, IVS1–5, IVS1–6, IVS1–110 and CD 39 in the HBB gene (region chr11: 5,226,658–5,227,244, Genome Browser GRCh38/hg38). All participants in this study signed the informed consent form approved by the research ethics committee of Clementino Fraga Filho University Hospital (protocol 5.737.364).
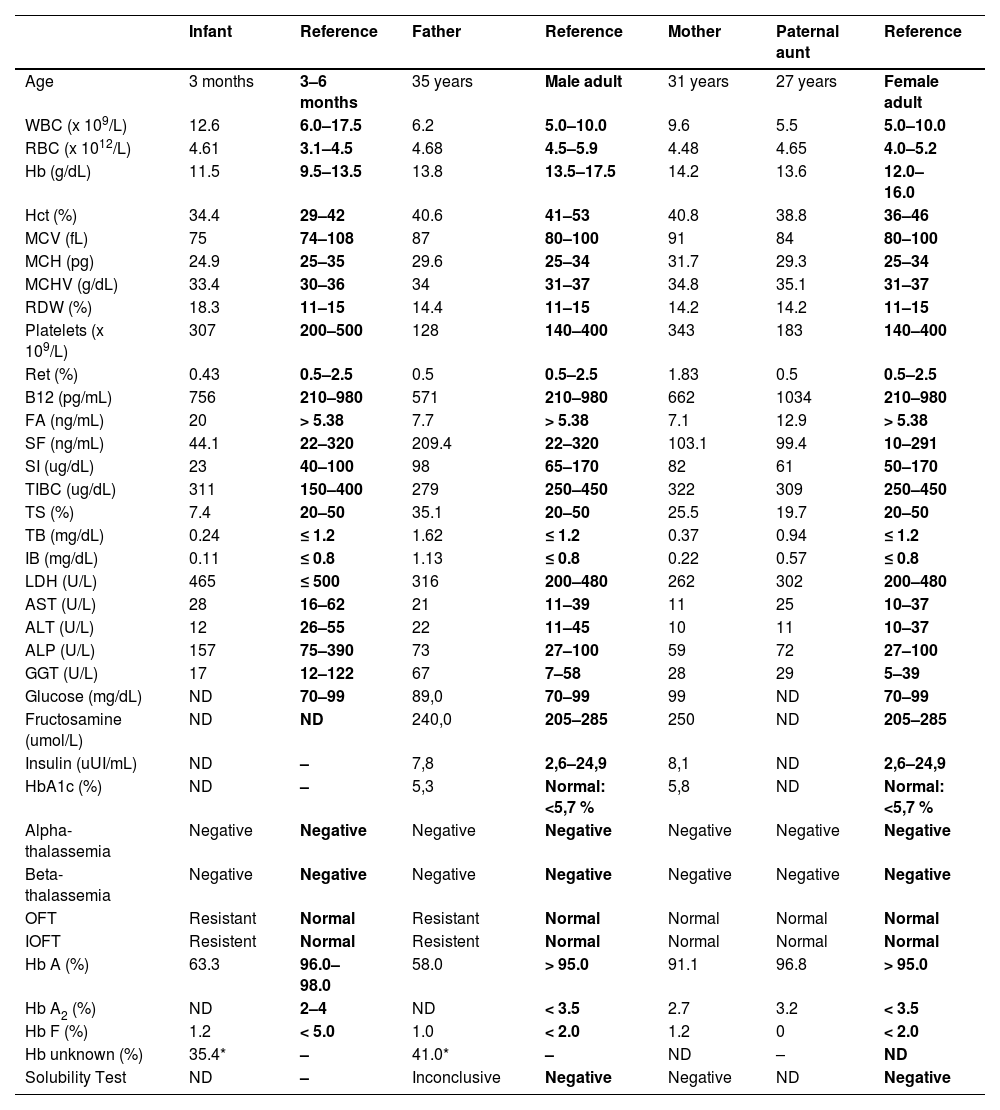
Case reportThe infant and his parents were referred to the Laboratory of Clinical Analysis of the Faculty of Pharmacy/UFRJ (LACFAR/UFRJ) to analyze a variant Hb detected in newborn screening tests for congenital diseases. The child and the father presented previous exams describing the presence of a variant Hb by Hb-HPLC, with a similar retention time to HbA2, in a single peak representing 35–40 % of total Hb. The case investigation was initiated with the hematological and biochemical examination of the child's father, mother, and paternal aunt. The infant's complete blood count presented a slight increase in RBC and diminished levels of MCH, whereas the RDW was increased (18 %), associated with an augmented resistance in the OFT. The evaluation of iron metabolism showed diminished serum levels of this ion and transferrin saturation index, indicating an iron deficiency anemia, which could influence RDW and OFT outcomes (Table 1). However, the investigation performed in the father's samples, bearing the same Hb variant, did not show iron deficiency but OFT resistance was maintained. Biochemical analysis revealed increased levels of unconjugated bilirubin and GGT, however with LDH and reticulocyte values between the healthy range, probably due to hepatic function impairment. The infant's erythrocyte hematoscopy revealed rare Echinocytes, Schistocytes and Elliptocytes, possibly caused by iron deficiency. In the father's hematoscopy, some Stomatocytes and the presence of rare macroplatelets were observed. The mother's HbA1c was slightly altered, above the reference value. The other laboratory evaluations of the family and molecular analysis of alpha and beta thalassemia, showed results within the reference value, as well as the Hb-HPLC profile (Table 1). Analysis of Hb-HPLC chromatograms was performed for the entire family for evaluation and comparison with previous data. The presence of 34.5 % variant Hb was confirmed for the infant, with a retention time (RT) of 3.82 s, superimposed on HbA2. In addition to the child, only the father showed variation in the Hb profile, with 41 % of the Hb variant and RT of 3.80 s (Figure 1A). Based on the chromatographic profile, percentage of variant Hb found and RT, candidate Hb were identified as HbG Copenhagen, HbE or Hb Maputo (BIORAD, https://hemoglobins.bio-rad.com/pages/chromatogrampage.aspx?plaid=2&metid=4). Then, alkaline Hb electrophoresis was performed, demonstrating similar migration to HbS, in heterozygosis (HbAS), for the father and the child, discarding HbE, which presents a distinct migration pattern (Figure 1B). Finally, the sequencing of a fragment of the beta-globin gene was carried out to identify the Hb variant. Figure 1C shows the sequencing electropherograms of the infant (Figure 1C.1), who presented the HBB:c.142G>T mutation in heterozygosis compatible with Hb Maputo, as well as his father. The laboratory parameters evaluated for the mother did not show any change when compared to the reference value, but the gene sequencing showed a mutation at position c.9T>C, which characterizes HBB:c.9TFigure 1C.2), already described for not promoting laboratory and clinical repercussions, being considered a silent mutation. It is noteworthy that the sequenced fragment discarded the presence of HbS, HbG Copenhagen and HbE for all investigated family members. Aunt did not show mutations in the analyzed fragment (data not shown).
Summary of laboratory data of the proband and his family members.
WBC: white blood cells; RBC: red blood cells; Hb: hemoglobin; Hct: hematocrit; MCV: mean corpuscular volume; MCH: mean corpuscular hemoglobin; MCHC: mean corpuscular hemoglobin concentration; RDW: red cell distribution width; Ret: reticulocyte count; B12: B12 Vitamin; FA: Folic Acid; SF: serum ferritin; SI: serum iron; TIBC: total iron binding capacity; TS: transferrin saturation; TB: total bilirubin; IB: indirect bilirubin; LDH: lactate dehydrogenase; AST: Aspartate Aminotransferase; ALT: Alanine Aminotransferase; ALP: Alkaline Phosphatase; GGT: Gamma-Glutamyltransferase; DM: Diabetes Mellitus; Alpha-thalassemia: α3.7kb, α4.2kb and α−SEA Delections; Beta-thalassemia: IVS1–1, IVS1–6, IVS1–110 and CD 39 mutations; OFT: Osmotic Fragility Test; IOFT: Incubated (37 °C/24 h) Osmotic Fragility Test; Hb A, Hb A2 and Hb F: Hemoglobins A, A2 and Fetal quantification by cationic exchange HPLC respectively. *Later identified as Hb Maputo. ND: not determined.
 Hb-HPLC of the infant showing unknown Hb with the same retention time of HbA2. (A.2) Hb-HPLC of the father showing Hb unknown Hb with the same retention time of HbA2. (A.3) Hb-HPLC of the mother showing normal profile; (B) Electrophoresis of hemoglobins at pH 8.0. 1: Hb A (mother); 2: Hb AS; 3: Hb A/Maputo (infant); 4: Hb SC; 5: Hb AS; 6: Hb A/Maputo (father). (C) Electropherogram of the globin gene: (C.1) Sequencing of the beta-globin gene revealed a mutation in the codon 47 (β47 Asp>Tyr-HBB: c.142G>T) in heterozygosis corresponding to Hb Maputo, (C.2) Sequencing of the beta-globin gene revealed a mutation in the codon 2 (β2 His>His; HBB: c.9T>C) in heterozygosis, a silent variant.")
(A.1) Hb-HPLC of the infant showing unknown Hb with the same retention time of HbA2. (A.2) Hb-HPLC of the father showing Hb unknown Hb with the same retention time of HbA2. (A.3) Hb-HPLC of the mother showing normal profile; (B) Electrophoresis of hemoglobins at pH 8.0. 1: Hb A (mother); 2: Hb AS; 3: Hb A/Maputo (infant); 4: Hb SC; 5: Hb AS; 6: Hb A/Maputo (father). (C) Electropherogram of the globin gene: (C.1) Sequencing of the beta-globin gene revealed a mutation in the codon 47 (β47 Asp>Tyr-HBB: c.142G>T) in heterozygosis corresponding to Hb Maputo, (C.2) Sequencing of the beta-globin gene revealed a mutation in the codon 2 (β2 His>His; HBB: c.9T>C) in heterozygosis, a silent variant.
The investigation of this clinical case began with the anomalous chromatographic profile of the infant and father, who had the Hb variant eluted together with HbA2 in concentrations greater than 35 %. According to Giambonal et al.,8 HbA2 levels greater than 8 % correspond to a variant Hb eluting at the same position. According to the characteristics presented by the laboratory tests, the possible candidates for variant Hb would be HbG Copenhagen, HbE or Hb Maputo. HbE was ruled out by the electrophoretic mobility of the variant Hb, similar to the mobility of HbS. Characteristics of a group of Hb known as “HbS like”,is that Hb has electrophoretic migration similar to the HbS pattern, but with different mutations. The solubility test has an inconclusive result: it does not have the characteristic precipitation of HbS, but it does not show a negative result. According to the database of globin variant genes (https://globin.bx.psu.edu/), both Hb Maputo and G Copenhagen present mutation in codon 47 of beta-globin, namely: HbG Copenhagen p.Asp47Asn HBB: c.142G>A and Hb Maputo p.Asp47Tyr HBB: c.142G>T. Only after sequencing, it was possible to identify the mutation present in heterozygosis in the beta-globin chain fragment of the child and his father, corresponding to Hb Maputo. In addition, the mother's electropherogram (Figure 01.C.2) showed a heterozygous mutation in codon 2 (HBB: c.9T>C p.His2His) corresponding to HBB:c.9T>C a silent mutation, without clinical repercussions and wild-type genotype for the Maputo variant. The paternal aunt did not present mutations in the analyzed fragment. Hb Maputo presents the substitution of the polar negatively charged amino acid Aspartic Acid by Tyrosine, with an aromatic and relatively non-polar side chain, at codon 47 of the beta-globin chain, located on the outside surface of the molecule.2 Literature data describe that Hb Maputo has alkaline electrophoretic mobility similar to HbS, in addition to normal molecular stability and affinity for oxygen.2 A fact that corroborates the scenario without clinical repercussions in the presence of this Hb variant, in heterozygosis, but the same cannot be said when in association with HbS. Marinucci et al. first described Hb Maputo in a 2-year-old child in association with HbS, who had severe anemia (Hb 8.5 g/dL), a palpable spleen and a marked clinical status, presenting hematoscopy with abnormal morphology in the red series (Target cell and Anisocytosis).2 A profile was also found in our investigated patient. In our findings, the father, who presented simple heterozygosity for Hb Maputo, did not present any clinical symptoms, which leads us to suggest that the presence of only this Hb variant in heterozygosity can be silently manifested. As the incidence of HbS is high in Brazil, it is crucial to identify and register Hb Maputo due to the risk of causing symptoms of severe anemia in individuals with associated genotypes (HbS/Hb Maputo).2 Iron deficiency anemia observed in the infant could favor resistance to hemolysis in the OFT, but the father did not present such nutritional deficiency and his result remained resistant, suggesting that the observed profile could be associated with the variant Hb. In hemoglobinopathies (thalassemia and HbS) and iron deficiency anemia, there is an increase in resistance to hemolysis due to the increased ability to incorporate a greater volume of water by red blood cells and remain intact in hypertonic solutions.9 This laboratory finding is interesting for Hb Maputo since it broadens the characterization of this hemoglobinopathy. Finally, the mother, who has a wild-type genotype for Hb Maputo and a heterozygote for HBB:c.9T>C, despite having normal glucose metabolism, the result for HbA1c was slightly above the reference value. Her other results were normal, including OFT, Hb-HPLC results and alkaline Hb electrophoresis. The HBB:c.9T>C mutation has been described to not effect on the quantification of HbA1c in ion exchange HPLC during the investigation of diabetic patients.7 Given the low frequency worldwide on Hb Maputo and the scarce existing literature and HBB: c. 9T>C, the findings of this family become relevant and can contribute to a better characterization of hemoglobin variants, helping to a better laboratory diagnosis and helping public health policies. Consequently, the expansion of the frequency record of rare Hb in our country sheds light on the possible association of that rare Hb with other Hb variants with high prevalence in Brazil. Despite this real possibility of interaction between Hb Maputo with other hemoglobin variants, only Hb Maputo has been previously described in association with HbS with severe anemia.2 Despite the association of mutations in HBB gene of Thalassemic patients with silent mutation HBB: c.9T>C no additional pathological effect was detected.6
This work was financially supported by Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), Brazil. We thank all the members of the Laboratory of Clinical Analysis of the School of Pharmacy (LACFar, UFRJ, Brazil) and HORIBA for their technical support.



