

Myeloproliferative neoplasms are caused by a clonal proliferation of a hematopoietic progenitor. First described in 1951 as ‘Myeloproliferative Diseases’ and reevaluated by the World Health Organization classification system in 2011, myeloproliferative neoplasms include polycythemia vera, essential thrombocythemia and primary myelofibrosis in a subgroup called breakpoint cluster region-Abelson fusion oncogene-negative neoplasms. According to World Health Organization regarding diagnosis criteria for myeloproliferative neoplasms, the presence of the JAK2 V617F mutation is considered the most important criterion in the diagnosis of breakpoint cluster region-Abelson fusion oncogene-negative neoplasms and is thus used as a clonal marker. The V617F mutation in the Janus kinase 2 (JAK2) gene produces an altered protein that constitutively activates the Janus kinase/signal transducers and activators of transcription pathway and other pathways downstream as a result of signal transducers and activators of transcription which are subsequently phosphorylated. This affects the expression of genes involved in the regulation of apoptosis and regulatory proteins and modifies the proliferation rate of hematopoietic stem cells.
Myeloproliferative neoplasms (MPNs) are clonal disorders of hematopoietic stem cells, in which there is an increased proliferation of the myeloid lineage with effective maturation resulting in peripheral blood leukocytosis, and increased red blood cell mass; these neoplasms can progress to fibrosis or leukemic transformation. The MPNs are a group of diseases that include polycythemia vera (PV), essential thrombocythemia (ET) and primary myelofibrosis (PMF), which are collectively known as breakpoint cluster region-Abelson fusion oncogene (BCR-ABL)-negative neoplasms. These three diseases share common clinical features, such as proliferation of hematopoietic stem cells independent of growth factors, bone marrow hypercellularity, increased risk of thrombotic events and hemorrhages.1–4
Studies carried out in 2005 identified abnormalities in the functioning of the Janus kinase 2 (JAK2) protein which is expressed by the JAK2 gene. Here, a sense mutation at exon 14 of the JAK2 gene (g.1849 G>T)5,6 results in the substitution of a valine for a phenylalanine at position 617 located in the pseudokinase JH2 domain. It is responsible for constitutively activating the JAK/signal transducers and activators of transcription (STAT) signaling pathway and other pathways downstream. The JAK2 V617F mutation occurs at a frequency of 95% in PV and of 55–60% in ET and PMF.5–10
Myeloproliferative neoplasmsDameshek1 carried out the first classification of Myeloproliferative Diseases in 1951. However, the World Health Organization (WHO) classification system published in 2011 introduced a new classification of tumors of hematopoietic and lymphoid tissue and changed the term to ‘myeloproliferative neoplasms’. The MPNs include chronic myeloid leukemia (CML), PV, PMF, ET, chronic neutrophilic leukemia (CNL), chronic eosinophilic leukemia (CEL), mast cell disease (MCD) and unclassified myeloproliferative neoplasms (MPN-u).11–13 CML is characterized by the presence of the Philadelphia chromosome (Ph) resulting from a translocation between chromosomes 9 and 22. The resulting hybrid gene, BCR-ABL, encodes a protein of 210kDa, displaying tyrosine kinase activity and affects growth and cell differentiation.14
In addition to their common features, PV is characterized primarily by an increase in red blood cell production, ET for excessive production of platelets, and PMF for morphological changes in megakaryocytes and the development of monocytes that lead to the secretion of angiogenic factors responsible for promoting fibrosis in the bone marrow.15
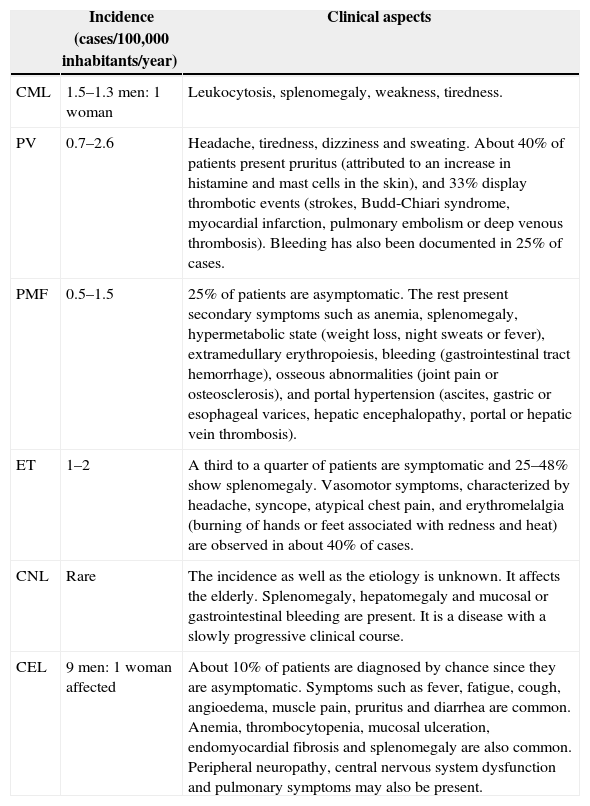
Table 1 presents the MPN types, the incidences and clinical aspects of each.
Myeloproliferative neoplasms.
| Incidence (cases/100,000 inhabitants/year) | Clinical aspects | |
|---|---|---|
| CML | 1.5–1.3 men: 1 woman | Leukocytosis, splenomegaly, weakness, tiredness. |
| PV | 0.7–2.6 | Headache, tiredness, dizziness and sweating. About 40% of patients present pruritus (attributed to an increase in histamine and mast cells in the skin), and 33% display thrombotic events (strokes, Budd-Chiari syndrome, myocardial infarction, pulmonary embolism or deep venous thrombosis). Bleeding has also been documented in 25% of cases. |
| PMF | 0.5–1.5 | 25% of patients are asymptomatic. The rest present secondary symptoms such as anemia, splenomegaly, hypermetabolic state (weight loss, night sweats or fever), extramedullary erythropoiesis, bleeding (gastrointestinal tract hemorrhage), osseous abnormalities (joint pain or osteosclerosis), and portal hypertension (ascites, gastric or esophageal varices, hepatic encephalopathy, portal or hepatic vein thrombosis). |
| ET | 1–2 | A third to a quarter of patients are symptomatic and 25–48% show splenomegaly. Vasomotor symptoms, characterized by headache, syncope, atypical chest pain, and erythromelalgia (burning of hands or feet associated with redness and heat) are observed in about 40% of cases. |
| CNL | Rare | The incidence as well as the etiology is unknown. It affects the elderly. Splenomegaly, hepatomegaly and mucosal or gastrointestinal bleeding are present. It is a disease with a slowly progressive clinical course. |
| CEL | 9 men: 1 woman affected | About 10% of patients are diagnosed by chance since they are asymptomatic. Symptoms such as fever, fatigue, cough, angioedema, muscle pain, pruritus and diarrhea are common. Anemia, thrombocytopenia, mucosal ulceration, endomyocardial fibrosis and splenomegaly are also common. Peripheral neuropathy, central nervous system dysfunction and pulmonary symptoms may also be present. |
In addition to the JAK2 gene, works carried out since 2005 have found other mutations in candidate genes, including the myeloproliferative leukemia (MPL), ten-eleven translocation 2 (TET2) a member of the TET oncogene family, additional sex comb-like 1 (ASXL1), Casitas B-lineage lymphoma (CBL), isocitrate dehydrogenase (IDH) and IKAROS family zinc finger 1 (IKZF1) genes, all identified within the group of BCR-ABL-negative neoplasms. Mutations in the JAK2 and MPL encoder genes seem to have greater effect and are more associated with PV, ET and PMF with frequencies around 95%, 55% and 60% for JAK2 and 0%, 0.3% and 10% for MPL.16
The functional consequences of these mutations in MPNs include disruption of the JAK/STAT signaling pathway, epigenetic modulation of transcription as well as abnormal accumulation of oncoproteins. However, it is still unclear how all these abnormalities contribute toward disease onset and clonal evolution of cells.16
Pathogenesis of myeloproliferative neoplasmsIn the literature, some evidence suggests that phenotypic characteristics observed in MPNs are caused due to disorders in the hematopoietic cell signaling process. It has been reported that hematopoietic progenitors are hypersensitive to several growth factors in PV, ET and PMF,17 and that abnormal myeloproliferation in patients with MPNs originates from constitutive activation of signal transduction pathways. These are caused by genetic rearrangements or mutations that affect tyrosine kinases or associated molecules.18 Based on this and other evidence, some research groups started to work with the hypothesis that alterations in the function of the JAK2 protein could be related to MPNs, since this protein is mainly responsible for the activation of molecules involved in cell signaling processes and red blood cell production.19
The Janus kinase 2 geneThe JAK2 gene, located on chromosome 9p24 in humans, includes 25 exons encoding a protein of about 1132 amino acids. This protein is named JAK2 and it is a member of the Janus kinase (JAK) family. Four members of this family have already been identified: JAK1, JAK2, JAK3 and Tyk2.20
In 2005, the discovery of the JAK2 mutation (JAK2 V617F) expanded the knowledge about the pathogenesis of BCR-ABL-negative neoplasms. A missense mutation at position g.1849 with a guanine-thymine transversion (g.1849 G>T)5,6 was identified in exon 14 of the JAK2 gene. This mutation results in the substitution of valine for phenylalanine at amino acid position p.617,5 affecting the pseudokinase domain (JH2) of the protein. The JAK2 V617F mutation is present in most patients with PV and in a subgroup of patients with ET and PMF (∼95% in PV, 50–60% in ET and 50–60% in PMF).5–9,19
It has been shown that the JAK2 V617F mutation causes genetic instability in gene expression by inducing changes in the chromatin structure and by reducing the apoptotic response to DNA damage. These are mechanisms that can increase the accumulation of genetic lesions leading to malignant transformations.12 Besides the g.1849 G>T (p.V617F) mutation,5,6 other mutations were found in exon 12 in 2007 namely: p.N542-543del, p.E543-D544del, p.K539L, p.F537-K539delinsL, p.H538-p.K539delinsL, p.H538QK539L, p.V536-1546dup11, p.F537-1546dup10+F547L, p.R541 E543delinsK-and-p.I540 E543delinsMK.21–23 These were found to show low frequencies of about 3–4% in PV patients. There are valid techniques that detect these mutations and their active search is justified not only due to the absence of the exon 14 mutation, but also in patients that have erythrocytosis and in those with low levels of erythropoietin.24
The Janus kinase 2 proteinMembers of the JAK2 protein family have seven homologous domains (JH) that are numbered from 1 to 7. The JH1 domain acts as a kinase domain, which contains the adenosine triphosphate (ATP)-binding and signaling pathway activation regions. The JH2 domain is a pseudokinase that is homologous to the JH1 domain. It lacks catalytic activity due to a substitution of an aspartate residue in the catalytic loop (His–Arg–Asp – HRD motif). JH2 has an inhibitory function that regulates both the basal activity of JAK and the cytokine-induced activation by interacting with the active site, JH1, blocking ATP and/or substrates binding to the catalytic site.25–27
Mutations in exon 12 of JAK2 are found in the binding region of the Src homology 2 (SH2) and JH2 domains. Although not directly located in the JH2 domain, these mutations may modify the structure of the pseudokinase domain, inducing erythropoietin hypersensitivity similar to that caused by the JAK2 p.V617F mutation.5,9,28,29
The Janus kinase 2/signal transducers and activators of transcription signaling pathwaySignaling receptorsCytokines from the hematopoietic system include interleukins (ILs), colony stimulating factors (CSFs), interferon (INF), erythropoietin (EPO) and thrombopoietin (TPO) all of which activate the signal transduction of the JAK/STAT signaling pathway.30,31 Most of these receptors are part of a type I-homodimer receptor family, that includes the erythropoietin (EPO-R) and thrombopoietin (TPO-R) receptors.32
The type I receptor is specifically expressed during erythroid and myeloid differentiation and its expression may account for the preferential activity of JAK2 V617F over myeloid lineage cells rather than lymphoid lineage cells, which lack this type of receptor.32
Janus kinasesJAKs are associated with the intracellular domains of cytokine receptors; they show intrinsic kinase activity mediated through their band-4.1 protein, ezrin, radixin, and moesin (FERM) and SH2 domains and are kept in an inactive state in the absence of specific receptor-activating ligands.33,34
Cytokine binding leads to a conformational change of the receptor, cytoplasmically affecting associated JAKs, causing activation and phosphorylation. Phosphorylated tyrosine residues in JAKs act as binding sites for the SH2 domains in signaling molecules. Mutations in JAK2 generate constitutive activation of the JAK/STAT pathway, mainly in STAT3 and STAT5. In addition to ‘hot spot’ mutations, constitutive activation of tyrosine kinases also occurs by chromosomal translocation, deletion and tandem duplication, which are common pathogenic events in hematopoietic malignancies.10,27,31–35
The STATs are located in the cytosol and migrate to the nucleus, regulating gene transcription only upon activation. Once associated with the phosphorylated sites of JAK, these STATs also phosphorylate and dimerize.26
Dimerized STATs migrate toward the nucleus, where they act as transcription factors, activating or repressing genes that are important in cell proliferation and survival. Some of these genes include those that express cyclins as well as anti-apoptotic proteins.36 STAT dimers also activate genes that encode inhibitory proteins that contribute toward the termination of cellular response. Some of these proteins bind to phosphorylated JAKs and inactivate them, as well as their associated receptors. Others bind to phosphorylated STAT dimers and stop them from binding to their target gene.37
JAK/STAT-mediated signaling control mechanisms include dephosphorylation of JAK and STAT. This is caused by an inhibition triggered by suppressors of cytokine signaling (SOCS) proteins, the lymphocyte adaptor protein (LNK also known as SH2B3) and CBL.30,32
Negative regulation of the Janus kinase 2/signal transducers and activators of transcription pathwayAs our knowledge about the JAK/STAT pathway has increased, important mechanisms for down-regulation of this pathway have been elucidated. These include removal of phosphates from cytokine receptors and activated STATs by tyrosine phosphatases, as well as by protein inhibitors of activated STAT (PIAS). PIAS inhibit transcriptional activation by binding and blocking access to target DNA. The next section will discuss three PIAS: SOCS, CBL and Lyme neuroborreliosis (LNB) proteins.
SOCS proteins are a family of cytokine signaling suppressors of the JAK/STAT pathway, characterized by the presence of an amino-terminal domain that is variable in size. It includes an inhibitory domain, a SH2 domain and a carboxy-terminal domain called SOCS box. Currently, there are eight members in this family, namely: SOCS1, SOCS2, SOCS3, SOCS4, SOCS5, SOCS6, SOCS7 and CIS. Under normal conditions, they are expressed at low levels in unstimulated cells while their expression increases when STATs are activated.30,31,38
SOCS can inhibit the JAK/STAT signaling through two mechanisms: (1) inhibition of JAK2 kinase activity by competing with the STATs SH2 domains for binding sites in the cytoplasmic domain of the receptor or (2) proteasomal degradation of signaling proteins by binding to the JH1 domain of JAK2, inhibiting the activation of all other JAK/STAT-associated pathways.39
Some mutations in different SOCS have been identified in MPNs, but are rarely found. Hypermethylation of CpG islands in SOCS1 and SOCS3 is associated with a decrease in the expression of these regulators in p.V617F-positive PV and ET patients.5,10
Other important components of this negative regulatory complex include multifunctional proteins with ubiquitin ligase activity, such as CBL. CBL is usually involved in the negative feedback of tyrosine kinase receptors by competitive blocking signaling. It induces proteasomal degradation of tyrosine kinases activated by ubiquitination. All CBL have single domain proteins that recognizes phosphorylated tyrosine residues that are present on activated tyrosine kinases.10,40
LNK, included in the SH2B protein family, negatively regulates the activity of JAK2 by binding to SH2-phosphorylated tyrosine. It also regulates the signaling of TPO-R and EPO-R, and it is detected in both JAK2 V617F-positive and -negative patients.10 Increased LNK deficiency increases the oncogenic ability of JAK2 to expand myeloid progenitors in vitro and in vivo.41
Mutation in the Janus kinase 2 gene and changes in protein functionIn addition to activating the JAK/STAT pathway, the p.V617F mutation5 also constitutively activates the rat sarcoma (RAS)/Mitogen-activated protein kinases (MAPK) and phosphoinositide 3-kinase (PI3K)/protein kinase B (PKB, aka Akt) pathways, which results in an increased expression of mitotic proteins, regulatory proteins from the cellular cycle [cyclins D1, CDC25A (cell division cycle)] and anti-apoptotic genes [B-cell lymphoma-extra-large (Bcl-XL) and Bcl2]. These abnormalities involving the JAK/STAT and related pathways result in proliferation of affected cells (Figure 1).10

Signaling pathways activated by cytokine receptor binding. PI3K, Akt and STAT are activated after the phosphorylation of JAK2. The active negative feedback regulatory proteins complete the activation pathways. Such control is ineffective in the presence of a mutation in the JH2 domain of JAK2. PI3K: phosphoinositide 3-kinase; Akt: protein kinase B; STAT: signal transducers and activators of transcription; JAK2: Janus kinase 2; EPO-R: erythropoietin receptor; SOCS: suppressors of cytokine signaling; Bcl-XL: B-cell lymphoma-extra-large; Myc: myelocytomatosis oncogene.
In mutated cells, down-regulation becomes inadequate and/or inefficient. SH2 domains, normally present in PIAS proteins, cannot dephosphorylate JAK2. In addition, the anti-apoptotic mechanisms are expressed at much higher levels than normal. An acquired mutation in one of these regulatory proteins can trigger an exacerbated activation of STAT, even in the absence of specific mutations in JAK2.42
Additional molecular events appear to interfere with the activity of JAK2, such as cytogenetics, which increase the kinase activity of the protein, the regulatory ability of phosphorylases and SOCS proteins, polymorphisms, and mutations in cytokine receptors.43
Questions about the different phenotypes in BCR-ABL-negative myeloproliferative neoplasms and the V617F mutationA further intriguing question for researchers regarding MPNs is how the same mutation can trigger three phenotypically different diseases. There is still no clear understanding of the molecular mechanisms involved in the process, just hypotheses. One of them is the dose–response gene hypothesis, which is associated with allele load. Another is that there may be epigenetic changes that predispose the patient's organism to develop certain MPNs as well as changes in genes specific in the development of malignancies, as well as mutations in other signaling pathways that might affect signaling in regulatory proteins.16
For Kralovics et al., Plo et al., Smith & Fan, Cross and Pagliarini,26,44–47 the phenotype of the disease is related to the concept of allelic load. Thus, low levels of mutant alleles would result in ET phenotype, while higher levels of mutated alleles would lead to a phenotype similar to PV. These higher levels of mutated alleles in hematopoietic stem cells could be associated with the patient's homozygous condition, caused by loss of heterozygosity, as explained by the theory of uniparental disomy.48
Presumably, homozygosity confers a proliferative advantage over other cells, which present only one mutated allele, increasing the activity level of the tyrosine kinase generated by the mutated JAK2. Patients with ET and PMF are mostly heterozygous and seldom show homozygosity.48
Besides the association between phenotype and homozygosity/heterozygosity, there are other hypotheses that have attempted to correlate the different phenotypes observed related to the JAK2 V617F mutation.
In their study, Plo et al.45 discuss the gene-dose hypothesis, which postulates that the JAK2 mutation can be a starting point for the three pathologies. They also discuss the existence of other genetic events that can modify the JAK2 kinase activity, which may explain the heterogeneity present in classic MPNs. Several mechanisms have been proposed to aid the understanding of loss of heterozygosity and/or the presence of new genetic abnormalities, including the double-stranded break repair mechanisms in DNA, homologous recombination and non-homologous end joining. All these lead to genomic instability, which highlights how essential the precise regulation of these mechanisms is for the maintenance of genome stability and diversity.49–51
According to Mascarenhas et al.52 biological events that lead to the initiation and progression of MPNs are linked not only to acquisition of genetic mutations such as JAK2 V617F. They are also due to epigenetic changes that do not affect the primary DNA sequence, but the gene expression responsible for chromatin remodeling. Deregulation of modulation processes between nucleosomes may result in silencing of tumor suppressor as well as differentiation genes, promoting cell survival by blocking apoptosis and senescence, contributing to malignant transformation. The researchers also highlighted two categories of epigenetic alterations in MPNs: (1) changes in the genes that encode proteins involved in remodeling of the chromatin structure, such as the TET2, ASXL1, JAK2 genes, among others, and (2) changes in the promoter site of genes essential for cell survival, differentiation and proliferation. Besides its role in signaling, the JAK2 V617F mutation also affects chromatin structure by blocking the recruitment of repressor proteins.9
Conflicts of interestThe authors declare no conflicts of interest.

