

To analyze the frequency of βS-globin haplotypes and alpha-thalassemia, and their influence on clinical manifestations and the hematological profile of children with sickle cell anemia.
MethodThe frequency of βS-globin haplotypes and alpha-thalassemia and any association with clinical and laboratorial manifestations were determined in 117 sickle cell anemia children aged 3–71 months. The confirmation of hemoglobin SS and determination of the haplotypes were achieved by polymerase chain reaction-restriction fragment length polymorphism, and alpha-thalassemia genotyping was by multiplex polymerase chain reaction (single-tube multiplex-polymerase chain reaction).
ResultsThe genotype distribution of haplotypes was 43 (36.7%) Central African Republic/Benin, 41 (35.0%) Central African Republic/Central African Republic, 20 (17.0%) Rare/atypical, and 13 (11.1%) Benin/Benin. The frequency of the α3.7 deletion was 1.71% as homozygous (−α3.7/−α3.7) and 11.9% as heterozygous (−α3.7/αα). The only significant association in respect to haplotypes was related to the mean corpuscular volume. The presence of alpha-thalassemia was significantly associated to decreases in mean corpuscular volume, mean corpuscular hemoglobin and reticulocyte count and to an increase in the red blood cell count. There were no significant associations of βS-globin haplotypes and alpha-thalassemia with clinical manifestations.
ConclusionsIn the study population, the frequency of alpha-thalassemia was similar to published data in Brazil with the Central African Republic haplotype being the most common, followed by the Benin haplotype. βS-globin haplotypes and interaction between alpha-thalassemia and sickle cell anemia did not influence fetal hemoglobin concentrations or the number of clinical manifestations.
Sickle cell anemia (Hb SS) is characterized by homozygosity for hemoglobin S (Hb S), consequent to a point mutation in the 6th codon of the β-globin gene on chromosome 11.1,2 It is the most prevalent hereditary hematologic disease in Brazil with elevated morbidity and mortality. It is estimated that 3500 children are born with Hb SS annually, one in every 1000 live births, whereas the frequency of heterozygotes in the general population is 2–8%.3,4 The clinical state of these patients is exceptionally variable, which may be influenced by different factors, such as the haplotypes of the βS-globin gene, the presence of alpha-thalassemia, and the fetal hemoglobin level (Hb F).1,2
Haplotypes of the βS-globin gene are determined by specific polymorphism patterns in the β gene complex. They originate from different regions of Africa, from which they receive their denominations: CAR or Bantu (Central African Republic), Benin (West Africa), Senegal (West Africa), Cameroon (West Africa) and Saudi-Indian (Asia, India and the Arabian Peninsula). Polymorphism sets not recognized by these classical patterns are referred to as atypical (ATP) and occur at a rate of 5–10%.5,6 In Brazil, due to the forced migration of African descendants, CAR and Benin (BEN) haplotypes are the most common.5,7–11
Alpha-thalassemia is the result of deficient synthesis of α globin chains in hemoglobin (Hb). The α chain genes are located on chromosome 16, and normal individuals have two α genes on each chromosome with this genotype being represented as αα/αα.12 The α3.7 deletion, which causes the most common form of alpha-thalassemia in Brazil,13 is the result of an unequal cross-over exhibiting a loss of a 3.7kb DNA fragment.14 Recent Brazilian studies show different prevalences for the α3.7 genotype according to the geographical ancestry of the individuals studied: 4.5% in European and 21.5% in African descendants,15 data similar to those obtained in populations of African descents by Sonati et al.16 Brazilian studies analyzing alpha-thalassemia in individuals with Hb SS present prevalences of α3.7 heterozygous individuals of from 17.6 to 28.2%, according to the geographic region.9–11,13,17
The interaction between Hb SS and alpha-thalassemia is associated with an inhibition of Hb S polymerization2,18,19 which decreases hemolytic episodes2,18 entailing reductions in mean corpuscular volume (MCV) and mean corpuscular hemoglobin (MCH) compared to Hb SS individuals without alpha-thalassemia.20 An improvement in the patients’ clinical state is observed with reductions in sickle cell crises.18,21 The presence of alpha-thalassemia is also significantly associated with a reduction in white blood cell (WBC)15 and reticulocyte2,17 counts, and increased levels of Hb A2.17,22–25 Although there is no defined relationship, increased levels of Hb F are also reported.2,24
The objective of this study was to assess the prevalence of βS-globin haplotypes and alpha-thalassemia in a group of children with Hb SS and their influence on hematological profile and clinical manifestations.
MethodsThis was a cross-sectional study with convenience sampling involving 117 of 122 under 6-year-old children with Hb SS, no matter the severity of the disease, attended at the Pediatric Hematology Outpatient Clinic of the Escola Paulista de Medicina, Universidade Federal de São Paulo. The samples were collected from September 2007 to December 2009; the age varied from 3 to 71 months (median age of 35.5 months) and 62 (52.99%) of the children were male. The study was approval by the Research Ethics Committee of the Escola Paulista de Medicina, Universidade Federal de São Paulo.
Exclusion criteriaPatients who had received red blood cell transfusions in the 3 months prior to sample collection and those receiving hydroxyurea were excluded from the study.
Clinical and laboratorial variablesClinical and laboratorial variables were obtained from the patient's medical records. The following laboratorial criteria were analyzed: Hb level, hematocrit (Ht), red blood cell (RBC), WBC, MCV, MCH, reticulocyte, and platelet counts, and Hb F levels.
The clinical variables were analyzed considering whether the patients had <3 or ≥3 types of manifestations per year including vaso-occlusive crisis (VOC), acute chest syndrome/pneumonia (ACS/PNM), infections (acute osteomyelitis and urinary tract infection) and episodes of acute splenic sequestration.
Molecular analysisDNA was extracted from WBCs in peripheral blood using phenol–chloroform extraction and ethanol precipitation.26 The Hb S mutation was confirmed by polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP).27 Single-tube multiplex-PCR was used for alpha-thalassemia genotyping using primers standardized by Chong et al.28 Determination of the haplotypes of the βs gene complex was performed by PCR-RFLP analysis using primers as proposed by Sutton et al.29 All tests were conducted using a thermal cycler from Veriti Applied Biosystems® (Foster City, CA, USA).
Statistical analysisNon-parametric tests were used for statistical analysis taking into account the variability and distribution of the study variables. Kruskal–Wallis with the Dunn test was used to compare more than two samples, and the Mann–Whitney test for two independent samples. Significance was set for alpha errors of 5% (p-value <0.05); statistically analysis was performed using the InStat-2 (GraphPad®, San Diego, CA, USA) and SigmaStat 3.11 software (Systat® Software, Chicago, IL, USA).
ResultsThe frequencies of haplotypes were 57.26% for CAR, 33.33% for BEN, 8.12% for ATP, 0.85% for SAUDI and 0.43% for Senegal. The genotype distribution was CAR/BEN in 43 (36.75%) patients, CAR/CAR in 41 (35.04%), Rare/ATP in 20 (17.09%) and BEN/BEN in 13 (11.11%). In the 20 Rare/ATP patients, the following combinations were identified: BEN/ATP in eight patients (6.84%), CAR/ATP in seven (5.98%), ATP/ATP in two (1.71%), and one individual (0.85%) with the SEN/CAR, SAUDI/CAR, and SAUDI/BEN haplotypes.
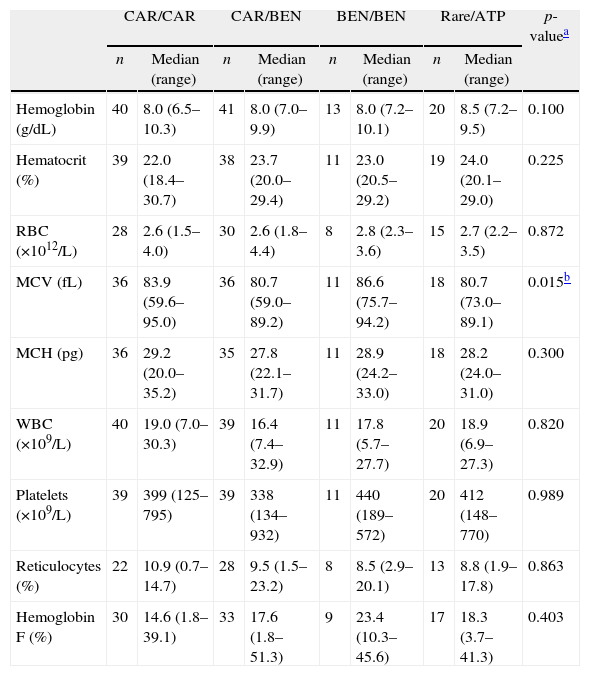
Even though the CAR/CAR group presented lower levels of Hb F than the other groups, no statistical difference was observed. MCV was different between the CAR/BEN and BEN/BEN haplotypes (p-value=0.01) (Table 1). A comparison between different haplotypes showed no difference concerning the number of clinical manifestations presented (p-value=0.22) (data not shown).
Hematological data according to βS-globin haplotype in 117 sickle cell anemia children.
| CAR/CAR | CAR/BEN | BEN/BEN | Rare/ATP | p-valuea | |||||
| n | Median (range) | n | Median (range) | n | Median (range) | n | Median (range) | ||
| Hemoglobin (g/dL) | 40 | 8.0 (6.5–10.3) | 41 | 8.0 (7.0–9.9) | 13 | 8.0 (7.2–10.1) | 20 | 8.5 (7.2–9.5) | 0.100 |
| Hematocrit (%) | 39 | 22.0 (18.4–30.7) | 38 | 23.7 (20.0–29.4) | 11 | 23.0 (20.5–29.2) | 19 | 24.0 (20.1–29.0) | 0.225 |
| RBC (×1012/L) | 28 | 2.6 (1.5–4.0) | 30 | 2.6 (1.8–4.4) | 8 | 2.8 (2.3–3.6) | 15 | 2.7 (2.2–3.5) | 0.872 |
| MCV (fL) | 36 | 83.9 (59.6–95.0) | 36 | 80.7 (59.0–89.2) | 11 | 86.6 (75.7–94.2) | 18 | 80.7 (73.0–89.1) | 0.015b |
| MCH (pg) | 36 | 29.2 (20.0–35.2) | 35 | 27.8 (22.1–31.7) | 11 | 28.9 (24.2–33.0) | 18 | 28.2 (24.0–31.0) | 0.300 |
| WBC (×109/L) | 40 | 19.0 (7.0–30.3) | 39 | 16.4 (7.4–32.9) | 11 | 17.8 (5.7–27.7) | 20 | 18.9 (6.9–27.3) | 0.820 |
| Platelets (×109/L) | 39 | 399 (125–795) | 39 | 338 (134–932) | 11 | 440 (189–572) | 20 | 412 (148–770) | 0.989 |
| Reticulocytes (%) | 22 | 10.9 (0.7–14.7) | 28 | 9.5 (1.5–23.2) | 8 | 8.5 (2.9–20.1) | 13 | 8.8 (1.9–17.8) | 0.863 |
| Hemoglobin F (%) | 30 | 14.6 (1.8–39.1) | 33 | 17.6 (1.8–51.3) | 9 | 23.4 (10.3–45.6) | 17 | 18.3 (3.7–41.3) | 0.403 |
CAR: Central African Republic; BEN: Benin; Rare: Senegal, and Saudi; ATP: atypical; RBC: red blood cells; MCV: mean corpuscular volume; MCH: mean corpuscular hemoglobin; WBC: white blood cells.
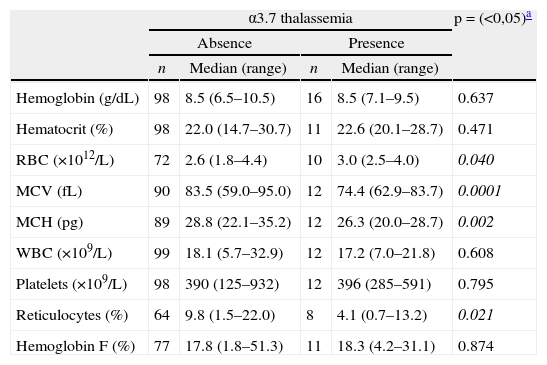
In relation to the occurrence of alpha-thalassemia, two patients (1.71%) were homozygous for the α3.7 deletion and 14 (11.97%) heterozygous, totaling 13.68% of alpha-thalassemia individuals (16/117). The alpha-thalassemia group (homozygotes and heterozygotes) presented significant reductions in MCV, MCH and reticulocyte counts, and had a significant increase in the RBC count compared to the group without the deletion. Although Hb F levels in the alpha-thalassemia group were higher in relation to the group without alpha-thalassemia (18.3%; range: 4.2–31.1 vs. 17.8%; range: 1.8–51.3, respectively) no significance was detected (p-value=0.84) (Table 2). Regarding the number of clinical manifestations, there was no significant difference between the groups (p-value=0.66).
Hematological data expressed as medians (range), according to presence of α3.7 thalassemia in 117 sickle cell anemia children.
| α3.7 thalassemia | p=(<0,05)a | ||||
| Absence | Presence | ||||
| n | Median (range) | n | Median (range) | ||
| Hemoglobin (g/dL) | 98 | 8.5 (6.5–10.5) | 16 | 8.5 (7.1–9.5) | 0.637 |
| Hematocrit (%) | 98 | 22.0 (14.7–30.7) | 11 | 22.6 (20.1–28.7) | 0.471 |
| RBC (×1012/L) | 72 | 2.6 (1.8–4.4) | 10 | 3.0 (2.5–4.0) | 0.040 |
| MCV (fL) | 90 | 83.5 (59.0–95.0) | 12 | 74.4 (62.9–83.7) | 0.0001 |
| MCH (pg) | 89 | 28.8 (22.1–35.2) | 12 | 26.3 (20.0–28.7) | 0.002 |
| WBC (×109/L) | 99 | 18.1 (5.7–32.9) | 12 | 17.2 (7.0–21.8) | 0.608 |
| Platelets (×109/L) | 98 | 390 (125–932) | 12 | 396 (285–591) | 0.795 |
| Reticulocytes (%) | 64 | 9.8 (1.5–22.0) | 8 | 4.1 (0.7–13.2) | 0.021 |
| Hemoglobin F (%) | 77 | 17.8 (1.8–51.3) | 11 | 18.3 (4.2–31.1) | 0.874 |
α3.7 thalassemia: heterozygous and homozygous; RBC: red blood cells; MCV: mean corpuscular volume; MCH: mean corpuscular hemoglobin; WBC: white blood cells.
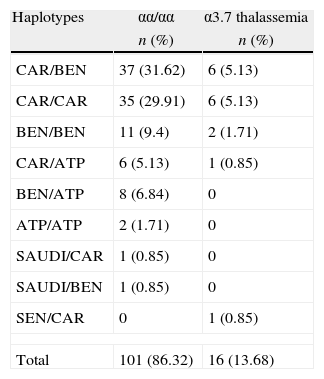
Table 3 shows the frequency of alpha-thalassemia in relation to the βS-globin haplotypes. Significant differences were observed in MCV values between individuals with and without alpha-thalassemia in the CAR/CAR and CAR/BEN groups (p-value=0.002 and 0.017, respectively) (Table 4).
Frequency of alpha-thalassemia according to βS-globin haplotypes in 117 children with sickle cell anemia.
| Haplotypes | αα/αα | α3.7 thalassemia |
| n (%) | n (%) | |
| CAR/BEN | 37 (31.62) | 6 (5.13) |
| CAR/CAR | 35 (29.91) | 6 (5.13) |
| BEN/BEN | 11 (9.4) | 2 (1.71) |
| CAR/ATP | 6 (5.13) | 1 (0.85) |
| BEN/ATP | 8 (6.84) | 0 |
| ATP/ATP | 2 (1.71) | 0 |
| SAUDI/CAR | 1 (0.85) | 0 |
| SAUDI/BEN | 1 (0.85) | 0 |
| SEN/CAR | 0 | 1 (0.85) |
| Total | 101 (86.32) | 16 (13.68) |
α3.7 thalassemia: homozygous and heterozygous; CAR: Central African Republic; BEN: Benin; SEN: Senegal; SAUDI: Asia, India and the Arabian Peninsula; ATP: atypical.
Mean corpuscular volume (MCV) presented as median (range) in CAR/CAR and CAR/BEN haplotypes according to the presence of α3.7 thalassemia.
| Haplotypes | α3.7 thalassemia | p-valuea | |||
| Absence | Presence | ||||
| n | Median (range) | n | Median (range) | ||
| CAR/CAR | 30 | 85.3 (68.5–95.0) | 6 | 77.4 (59.6–79.6) | 0.002 |
| CAR/BEN | 32 | 81.4 (59.0–89.2) | 3 | 71.1 (67.2–75.1) | 0.017 |
α3.7 thalassemia: homozygous and heterozygous; CAR: Central African Republic; BEN: Benin.
βS-globin gene haplotypes are being studied in Brazil and in other countries with the objective of determining their frequency in different regions and their influence on the clinical manifestations of Hb SS, since this is not yet well characterized.2 Alpha-thalassemia has also been considered an important modifying factor of clinical severity in Hb SS as it reduces hemolytic episodes and alters the hematological profile of patients.18
In patients with Hb SS, the elevated concentration of Hb F protects against vaso-occlusive events, consequently reducing clinical manifestations. Steinberg30 reported that Hb F is the most powerful disease modifier and Powars1 stated that, in being related to low Hb F levels, the CAR haplotype is associated with more severe clinical manifestations, while SEN and SAUDI haplotypes present high Hb F levels with milder clinical manifestations, and the BEN haplotype presents intermediate Hb F levels and clinical course.
In the current study, the CAR haplotype was the most common haplotype encountered, followed by BEN; these data are similar to other studies conducted in the southeast of Brazil.5,7–11 Despite the fact that there was no significant difference in Hb F levels among haplotypes, these values follow the pattern described in the literature, that is, lower values in CAR/CAR and CAR/BEN genotypes, followed by BEN/BEN.7,10
No significant difference was observed in the number of clinical manifestations between the haplotype types, probably due to the fact that the studied population presented a mean age of 37 months, when Hb F levels are still elevated in Hb SS individuals,31 and many clinical manifestations are not apparent at this age. Another possible factor might be the miscegenation of the Brazilian population and consequent elevated number of individuals with heterozygous haplotypes. A study conducted by Silva Filho et al.11 with a similar age group, encountered similar results.
The Rare/ATP group consists of different haplotype combinations, mostly involving CAR haplotype heterozygotes (11/25=44%) rather than heterozygotes and homozygotes with the ATP haplotype. According to Zago et al., 6 ATP haplotypes can be considered a variation of the Bantu haplotype, and thus we can suppose that they might be related to greater severity of the disease. Although the rare haplotypes (SEN and SAUDI) are associated with a milder clinical course, they did not influence the results obtained as they were identified in only three patients, confirming their low prevalence in the study region.7
Some hematological parameters are related to the severity of the disease and are directly implicated in the physiopathology of Hb SS, hence the importance of assessing their association with haplotypes and alpha-thalassemia. In the comparison between the different haplotype groups, the CAR/CAR group presented significantly lower Hb levels, followed by the CAR/BEN. These data are similar to results by Silva Filho et al.11 and de Neonato et al.24
Low MCV and MCH values are associated with a reduction in the synthesis of α chains and related to the effect that alpha-thalassemia exerts on the polymerization of Hb S and on the deformity of sickled cells, making them less dense.20 Other than this, there is evidence that microcytosis, per se, exert beneficial effects on vaso-occlusive complications in Hb SS.2,21
The frequency of alpha-thalassemia in the study population was similar to that found in other Brazilian studies performed on patients with Hb SS.9,10,17
Hb values did not differ between the groups with or without alpha-thalassemia, similar to the results reported by Silva Filho et al.11 and Stevens et al.23 Significant reductions in MCV and MCH levels and reticulocyte counts, and a significant increase in the RBC count in α3.7 deletion patients confirms data in the literature.2,17,22 There was no statistically significant difference in the WBC count between the groups, contrary to other publications that reported a reduction in the WBC count in individuals with alpha-thalassemia.17,22
Hb F levels did not show any significant difference between the two groups, similar to results obtained by Silva Filho et al.,11 Belisário et al.17 and Mouélé et al.,22 in studies conducted with similar age groups.
The current study did not observed any influence of alpha-thalassemia on clinical manifestations, different to other studies where the presence of alpha-thalassemia preserved splenic function, leading to persistence of splenomegaly in patients with homozygous α3.7 deletion.31 The fact that Hb F is still elevated in the study group might also justify the lack of influence of the deletion in alpha-thalassemia on the clinical manifestations of these patients.
One factor that might be considered as a bias in this study is that data collection of clinical events was from the patients’ medical records, which could be considered deficient. However the patients were followed-up in the institution, not only in the outpatient clinic but also in the emergency ward and during hospitalization, which reduces or discards bias caused by dependence on the memory of the patient's family.
ConclusionsThe frequency of alpha-thalassemia in this study was similar to previous studies in Brazil, with the CAR haplotype being the most common, followed by the BEN haplotype. The βS-globin gene haplotypes and the presence of alpha-thalassemia did not influence the levels of Hb F or the number of clinical manifestations presented. We believe that a prospective follow-up of these patients will provide more knowledge on the influence of alpha-thalassemia and the βS-globin gene haplotypes on clinical manifestations in Hb SS.
Conflicts of interestThe authors declare no conflicts of interest.



