

Haptoglobin genotypes, and interleukin-6 and -8 participate in the pathophysiology of sickle cell anemia. The expression of cytokines is regulated by genetic mechanisms however the effect of haptoglobin polymorphisms on these cytokines is not fully understood. This study aimed to compare the frequency of haptoglobin genotypes and the interleukin-6 and -8 concentrations in sickle cell anemia patients and controls to investigate the association between haptoglobin genotypes and cytokine levels.
MethodsSixty sickle cell anemia patients and 74 healthy individuals were analyzed. Haptoglobin genotypes were determined by multiplex polymerase chain reaction, and the interleukin-6 and -8 levels by enzyme linked immunosorbent assay. The association between haptoglobin genotypes and cytokines was investigated by statistical tests.
ResultsHp2-1 was the most common genotype in both the cases and controls while Hp1-1 was less frequent among sickle cell anemia patients. Interleukin-6 and -8 levels were higher in patients than controls (p-value <0.0001). There was no significant difference in interleukin-6 and -8 concentrations between the genotypes (p-value >0.05). A similar trend was observed among the controls.
ConclusionAlthough, levels of interleukin-6 and -8 were higher in the sickle cell anemia patients, they appeared not to be related to the haptoglobin genotypes. Further investigations are necessary to identify factors responsible for increased secretion of the interleukin-6 and -8 pro-inflammatory cytokines in patients with sickle cell anemia.
Sickle cell anemia (SCA) is the most common hereditary monogenic disease in Brazil, where the prevalence of the trait in the general population is 2–8% and the estimated number of affected individuals is about 7.2 million.1 Sickle hemoglobin (Hb S) is caused by a mutation of a single nucleotide (HBB glu(E)6Val(A); GAG>GTG; rs334; HBB:c.20A>T; MIM: #603903)2 in the beta globin gene, which results in glutamic acid being substituted by valine at position 6 of the beta globin chain. This change leads to Hb S polymerization, especially under low oxygen tension.3
Patients with SCA suffer from acute and chronic vascular occlusion due to polymerization of Hb S in red cells. Vascular occlusion is a major pathophysiologic event in SCA, leading to variety of morbidities such as painful crisis, acute chest syndrome, stroke, aseptic necrosis of bones, priapism, leg ulcers and proliferative retinopathy. Patients also experience chronic hemolytic anemia, inflammation, cell adhesion, tissue hypoxia, organ ischemia and tissue infarction.4
Hemolytic anemia, a major complication of SCA is linked to reduced nitric oxide (NO) bioavailability. In normal conditions, NO binds to soluble guanylate cyclase, which converts guanosine triphosphate to cyclic guanosine monophosphate. It causes relaxation of vascular smooth muscle and eventual peripheral vasodilatation. Plasma hemoglobin released from intravascular hemolysis of sickled red cells consumes NO and impairs its homeostatic vascular functions. Apart from the scavenging effects of cell-free plasma Hb on NO, reactive oxygen species (ROS) also scavenge NO and deplete its pool. The generation of ROS in patients with sickle cell disease (SCD) results from tissue ischemia that occurs during vaso-occlusion and the accompanying ischemia-reperfusion injury.5 It has also been suggested that the plasma level of l-arginine, a precursor of NO, is depleted during severe hemolysis, vaso-occlusive crises and acute chest syndrome. This depletion is partly due to increased levels of plasma arginase in these conditions, especially during severe hemolysis.6
Increased expression of adhesion molecules, including interleukin (IL)-6 also stimulates the adherence of red cells and neutrophils to the endothelium.4,7 IL-6, a multifunctional cytokine, plays a strategic role in host defense and exhibits both pro-inflammatory and anti-inflammatory properties. It is encoded by the IL-6 gene which is located on chromosome 7 at 7p21-p14, between D75135 and D75370.8 Apart from stimulation of B cell proliferation into plasma cells, its other biological activities include induction of the secretion of acute phase proteins by liver cells, T-cell activation and differentiation into cytotoxic cells, and activation of hematopoiesis.9
Several studies have shown that the levels of cytokines in patients with SCA, even in steady state, are elevated.10–13 In individuals with SCA, pro-inflammatory cytokines such as IL-1β, IL-6, IL-8 and tumor necrosis factor alpha (TNF-α) cause chronic endothelium activation and adhesion of sickled red cells, and are thus responsible for the constant local tissue ischemia and necrosis seen. In addition, there is increased leukocyte aggregation in response to inflammatory cytokines in SCA.10
The primary mechanism of defense against the deleterious effects of free Hb is provided by haptoglobin (Hp), an acute phase α2-globulin glycoprotein and important physiologic antioxidant, which binds to plasma Hb forming a soluble complex (Hb-Hp complex) to eliminate Hb from the plasma. This clearance is important because the complex is not inert and can catalyze oxygen reactive species and consume NO.14–16
The Hp locus in the chromosome, 16q22, is polymorphic. Two classes of alleles have been identified: 1 and 2, and the Hp-2 allele presents a duplicated DNA segment of 1700 base pairs (bp) due to an intragenic duplication of exons 3 and 4.17,18 In South America and Africa, the Hp-1 allele predominates, unlike in Southeast Asia where the frequency of the Hp-1 allele is lower than the Hp-2 allele.19 There are three possible genotypes for the Hp in humans: Hp 1-1, Hp 2-1 and Hp 2-2.
These phenotypes differ in the structure of the protein, electrophoretic mobility, antioxidant and anti-inflammatory properties, hence they exhibit different clinical effects.18,20,21Hp 1-1 is the most biologically active in binding free Hb and suppressing consequent inflammatory responses. Hp 2-2 is the least biologically active with a reduced overall activity and Hb clearance effects, while Hp 2-1 is intermediate.14 Hp significantly reduces the synthesis of ROS in individuals with the Hp 1-1 genotype because of its potent antioxidant activity, but in people with the Hp 2-2 polymorphism, the antioxidant activity is weak which ultimately favors the persistence of inflammatory response.22,23 Guetta et al. and Uskokovic et al. reported that the Hp 1 allele when bound to free Hb, stimulates the secretion of IL-6 and IL-10 more significantly than the Hp 2 allele.14,24
In patients with SCA, a disease characterized by both chronic and acute inflammatory and hemolytic states, the modulatory role of the Hp genotype on the expression of these cytokines is still largely understudied. The present study was therefore designed to investigate the association between the different Hp polymorphisms and the secretion of IL-6 and IL-8 in Brazilian SCA patients in comparison to healthy individuals from the same locality.
MethodsCases and controlsThis case control study included 60 patients with SCA, aged 13 years and above, who were recruited from the Hereditary Anemia Outpatient Clinic of the Hematology and Blood Transfusion Unit, Universidade Federal de São Paulo (UNIFESP), Brazil. At the time of recruitment, all the patients were in steady state, that is, they had had no acute event for at least twelve consecutive weeks. The control group consisted of 74 apparently healthy, age and gender-matched Hb AA volunteers from the same locality.
Pregnant women, individuals with acute or chronic infections, chronic renal impairment, rheumatologic disorders, patients on hydroxyurea therapy, chronic blood transfusion therapy and those who had had a blood transfusion within the preceding three months were excluded from the study. All participants or their parents/guardians (in the cases of adolescents) gave written informed consent before participation in the study. Moreover, permission for the study was obtained from the Research Ethics Committee of the institution.
Sociodemographic characteristics of the study participants were obtained using a structured questionnaire while data on SCA complications were retrieved from medical charts.
Determination of plasma level of cytokinesWhole blood samples were collected from antecubital veins under minimal tourniquet pressure. The plasma levels of IL-6 and IL-8 were determined using high sensitivity enzyme linked immunosorbent assays (ELISA) according to the instructions provided by the manufacturer. The IL-6 ELISA (BD OptEIA, BD Biosciences, San Diego, USA) had a normal range 2.2–12.1pg/mL and limit of detection of 2.2pg/mL. For the IL-8 ELISA (BD OptEIA, BD Biosciences, San Diego, USA), the normal range was 10.2–34.3pg/mL and the limit of detection was 0.8pg/mL.
DNA extractionBuffy-coat samples, which were stored at −80°C at the Molecular Biology Laboratory of UNIFESP, were defrosted and subjected to red blood cell lysis with saponin. Leukocytes were washed with Solution A (100mM KCl, 10mM Tris and 2.5mM MgCl2). DNA extraction was carried out using Solution A without saponin and Solution B (10mM Tris, 2.5mM MgCl2, 1% Tween-20, 1% Triton ×100 and proteinase K). After one-hour incubation at 60°C, DNA was purified with Phenol:chloroform:isoamyl alcohol (25:24:1).25
Haptoglobin genotypingHp genotyping was performed by a polymerase chain reaction (PCR) technique as described by Koch et al. in 2002.17 The method consists of two protocols. In protocol 1, the 1757bp allele 1 specific sequence and the 3481bp allele 2 specific sequence were amplified. In homozygous individuals, the intensity of the 1757bp allele-1 specific band is higher than the 3481bp allele-2 specific band. For 22 patients with SCA, it was not possible to define conclusively whether the 3481bp band was present. These samples were submitted to protocol 2 in order to amplify the 349bp allele-2 specific sequence. Protocol 2 involved the amplification of the 349bp allele 2 specific sequence to confirm when the result of the first assay was not conclusive. The PCR products were separated by agarose gel electrophoresis.
Statistical analysisData was analyzed using the Statistical Package for the Social Sciences (SPSS) computer program version 17.0. The mean plasma values of the interleukins were compared between cases and control by the independent sample t-test, while IL values between the HP 1-1, Hp 2-1 and Hp 2-2 genotypes were compared with analysis of variance (ANOVA). The frequencies of Hp polymorphisms in the patients and the controls were compared with the chi-square test. Statistical significance was set for p-values <0.05.
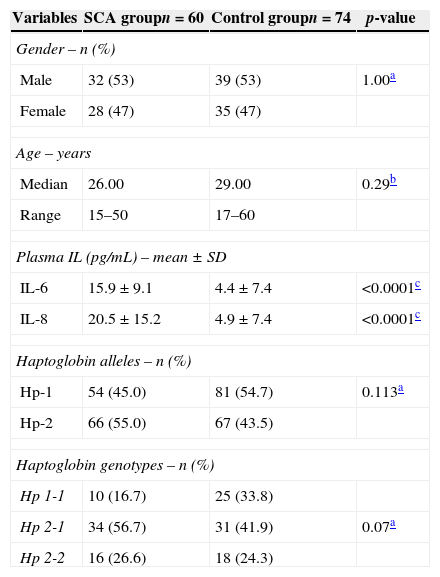
ResultsAs shown in Table 1, the age and gender profiles were similar between SCA patients and the control group.
Sociodemographic, haptoglobin genotypes and plasma levels of interleukin-6 and -8 between the sickle cell anemia patients and controls.
| Variables | SCA groupn=60 | Control groupn=74 | p-value |
|---|---|---|---|
| Gender – n (%) | |||
| Male | 32 (53) | 39 (53) | 1.00a |
| Female | 28 (47) | 35 (47) | |
| Age – years | |||
| Median | 26.00 | 29.00 | 0.29b |
| Range | 15–50 | 17–60 | |
| Plasma IL (pg/mL) – mean±SD | |||
| IL-6 | 15.9±9.1 | 4.4±7.4 | <0.0001c |
| IL-8 | 20.5±15.2 | 4.9±7.4 | <0.0001c |
| Haptoglobin alleles – n (%) | |||
| Hp-1 | 54 (45.0) | 81 (54.7) | 0.113a |
| Hp-2 | 66 (55.0) | 67 (43.5) | |
| Haptoglobin genotypes – n (%) | |||
| Hp 1-1 | 10 (16.7) | 25 (33.8) | |
| Hp 2-1 | 34 (56.7) | 31 (41.9) | 0.07a |
| Hp 2-2 | 16 (26.6) | 18 (24.3) | |
SCA: sickle cell anemia; SD: standard deviation, IL: interleukin.
The plasma levels of IL-6 and IL-8 were significantly higher in patients compared to the controls (p-value <0.0001) as shown in Table 1. There was about a four-fold increase in the levels of the plasma IL-6 and IL-8 in patients with SCA compared to the healthy controls.
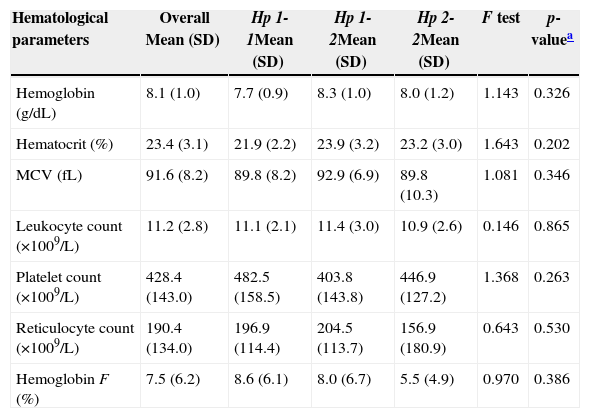
Haptoglobin alleles and genotypesThe frequencies of Hp-1 alleles among the cases and controls were 54 (45.0%) and 81 (54.7%) respectively (Table 1). Hp 2-1 was the most common Hp genotype in cases and controls, accounting for 34 (56.7%) and 31 (41.9%) respectively. The least common genotype in SCA patients was Hp 1-1, 16.7%. There was no statistically significant difference in the frequency distribution of Hp genotypes between cases and the controls [χ2=5.280, degree of freedom (df)=2, p-value=0.07]. The mean values of all the hematological parameters were not significantly different between the three Hp genotype groups (Table 2).
Hematological profile of the 60 sickle cell patients.
| Hematological parameters | Overall Mean (SD) | Hp 1-1Mean (SD) | Hp 1-2Mean (SD) | Hp 2-2Mean (SD) | F test | p-valuea |
|---|---|---|---|---|---|---|
| Hemoglobin (g/dL) | 8.1 (1.0) | 7.7 (0.9) | 8.3 (1.0) | 8.0 (1.2) | 1.143 | 0.326 |
| Hematocrit (%) | 23.4 (3.1) | 21.9 (2.2) | 23.9 (3.2) | 23.2 (3.0) | 1.643 | 0.202 |
| MCV (fL) | 91.6 (8.2) | 89.8 (8.2) | 92.9 (6.9) | 89.8 (10.3) | 1.081 | 0.346 |
| Leukocyte count (×1009/L) | 11.2 (2.8) | 11.1 (2.1) | 11.4 (3.0) | 10.9 (2.6) | 0.146 | 0.865 |
| Platelet count (×1009/L) | 428.4 (143.0) | 482.5 (158.5) | 403.8 (143.8) | 446.9 (127.2) | 1.368 | 0.263 |
| Reticulocyte count (×1009/L) | 190.4 (134.0) | 196.9 (114.4) | 204.5 (113.7) | 156.9 (180.9) | 0.643 | 0.530 |
| Hemoglobin F (%) | 7.5 (6.2) | 8.6 (6.1) | 8.0 (6.7) | 5.5 (4.9) | 0.970 | 0.386 |
MCV: mean corpuscular volume.
Table 3 compares the mean plasma concentrations of IL-6 of the three Hp genotypes. For the cases, there was no significant difference in the mean plasma concentrations of IL-6 for the Hp 1-1, Hp 2-1 and Hp 2-2 genotypes (14.6; 16.5 and 15.4pg/mL, respectively; F=84.47; p-value=0.54). For the controls, the difference in the mean plasma concentrations of IL-6 for the Hp genotype groups was also similar (F=53.23; p-value=0.161).
Association between haptoglobin genotypes and plasma concentrations of interleukin-6 and -8 levels.
| IL-6 concentration (pg/mL) | Hp1-1 | Hp1-2 | Hp2-2 | p-value |
|---|---|---|---|---|
| Patients | ||||
| Mean | 14.6 | 16.5 | 15.4 | 0.538b |
| Standard deviation | 3.2 | 11.0 | 6.9 | |
| Median | 14.7 | 13.4 | 15.3 | |
| Range | 10.0–20.9 | 2.2–53.8 | 2.2–30.4 | |
| Controls | ||||
| Mean | 2.5 | 4.6 | 6.8 | 0.161b |
| Standard deviation | 1.5 | 8.5 | 9.5 | |
| Median | 2.2 | 2.2 | 2.2 | |
| Range | 2.2–9.5 | 2.2–48.5 | 2.1–32.1 | |
| p-value | <0.0001a | <0.0001a | 0.0061a | |
| IL-8 concentration (pg/mL) | Hp1-1 | Hp1-2 | Hp2-2 | p-value |
|---|---|---|---|---|
| Patients | ||||
| Mean | 20.1 | 19.2 | 23.5 | 0.522b |
| Standard deviation | 10.5 | 14.5 | 19.1 | |
| Median | 18.3 | 16.6 | 17.7 | |
| Range | 2.6–38.9 | 1.4–53.5 | 1.6–68.3 | |
| Controls | ||||
| Mean | 4.8 | 5.7 | 3.5 | 0.606b |
| Standard deviation | 5.9 | 9.0 | 6.7 | |
| Median | 2.7 | 1.1 | 1.0 | |
| Range | 0.8–19.6 | 0.3–37.5 | 0.1–28.5 | |
| p-value | 0.0002a | <0.0001a | <0.0001a | |
IL: Interleukin.
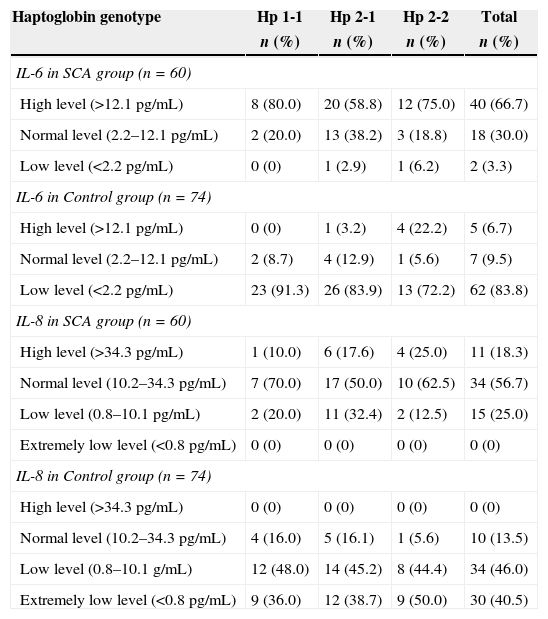
Two-thirds of patients with SCA, comprising of 8 with Hp 1-1; 20 with Hp 2-1 and 12 with Hp 2-2 had high levels of IL-6 i.e. IL-6 >12.1pg/mL, while only two (3.3%) had low levels (values <2.2pg/mL) (Table 4). However, among the controls, the majority, 62 (83.8%) had low levels of IL-6.
Relationship between haptoglobin genotypes and interleukin-6 and -8 levels.
| Haptoglobin genotype | Hp 1-1 | Hp 2-1 | Hp 2-2 | Total |
|---|---|---|---|---|
| n (%) | n (%) | n (%) | n (%) | |
| IL-6 in SCA group (n=60) | ||||
| High level (>12.1pg/mL) | 8 (80.0) | 20 (58.8) | 12 (75.0) | 40 (66.7) |
| Normal level (2.2–12.1pg/mL) | 2 (20.0) | 13 (38.2) | 3 (18.8) | 18 (30.0) |
| Low level (<2.2pg/mL) | 0 (0) | 1 (2.9) | 1 (6.2) | 2 (3.3) |
| IL-6 in Control group (n=74) | ||||
| High level (>12.1pg/mL) | 0 (0) | 1 (3.2) | 4 (22.2) | 5 (6.7) |
| Normal level (2.2–12.1pg/mL) | 2 (8.7) | 4 (12.9) | 1 (5.6) | 7 (9.5) |
| Low level (<2.2pg/mL) | 23 (91.3) | 26 (83.9) | 13 (72.2) | 62 (83.8) |
| IL-8 in SCA group (n=60) | ||||
| High level (>34.3pg/mL) | 1 (10.0) | 6 (17.6) | 4 (25.0) | 11 (18.3) |
| Normal level (10.2–34.3pg/mL) | 7 (70.0) | 17 (50.0) | 10 (62.5) | 34 (56.7) |
| Low level (0.8–10.1pg/mL) | 2 (20.0) | 11 (32.4) | 2 (12.5) | 15 (25.0) |
| Extremely low level (<0.8pg/mL) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| IL-8 in Control group (n=74) | ||||
| High level (>34.3pg/mL) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Normal level (10.2–34.3pg/mL) | 4 (16.0) | 5 (16.1) | 1 (5.6) | 10 (13.5) |
| Low level (0.8–10.1g/mL) | 12 (48.0) | 14 (45.2) | 8 (44.4) | 34 (46.0) |
| Extremely low level (<0.8pg/mL) | 9 (36.0) | 12 (38.7) | 9 (50.0) | 30 (40.5) |
SCA: sickle cell anemia.
Furthermore, the mean plasma values of IL-8 of the three Hp genotypes were similar both for the cases and the controls (F=97.34; p-value=0.52 and F=56.82; p-value=0.61, respectively) (Table 3).
As shown in Table 4, the majority of the cases (56.7%) as well as the controls (46.0%) had normal levels of plasma IL-8.
DiscussionSCA is a monogenic disease with an exceptional phenotypic variability that is regulated by several known and unknown genetic factors. Understanding the vascular and inflammatory components of the disease can help in providing information on the possible genomic sites that influence the phenotype. Studies on the Hp polymorphisms, an important marker of vascular disease and pro-inflammatory cytokines may therefore provide useful information. To the best knowledge of the authors, this is the first study that examined the relationship between Hp polymorphisms and interleukins among Brazilian SCA patients. The ability to predict the phenotype of SCA during the first months of life or even in the prenatal period will allow a more precise prognosis and individualized treatment.
Intravascular hemolysis produces a state of endothelial dysfunction, vascular proliferation, inflammation and oxidative stress. Recent studies have demonstrated the important roles of free plasma Hb in reducing NO. Intravascular hemolysis and the reduction of NO causes increased expression of endothelin-1, and activation of endothelial adhesion molecules and platelets. Thus, not only do they cause vasoconstriction, but NO is involved in endothelial activation and proliferation that eventually contributes to the pathogenesis of SCA.4,26 It is known that sickled red cells bind to the vascular endothelium cells by integrin alpha-4 beta-1. This membrane protein then acts as a receptor for either fibronectin or VCAM-1. In the endothelium, VCAM-1 is stimulated by inflammatory cytokines, such as IL-6 and IL-8 which are released by the activated leucocytes.27,28 Heme and hemin, which are released into the circulation during the active phase of sickle-cell disease, also contribute to inflammatory states in individuals with SCA by increasing the expression of endothelial adhesion molecules and adherence of leukocytes and reticulocytes to endothelial cells.29
In the present study, higher levels of both IL-6 and IL-8 were detected in SCA individuals compared to the control group (p-value <0.001). Our data agree with previous studies that identified elevated levels of inflammatory cytokines in SCA patients.10,30,31
Considering that the transcription of cytokines is regulated by a great number of genetic mechanisms, this study intended to verify whether the Hp polymorphisms influence cytokine secretion. Hp has emerged as one of the most important SCD phenotypic modulators, as it is a protein with high capability to bind to free Hb in the plasma, forming the Hp–Hb complex and thereby preventing heme-catalyzed oxidative damage.32 Although the three Hp phenotypes have the same ability to bind to free Hb, the speed of heme release can be related to differences in their molecular size. Hp 2-2, which is a more complex polypeptide, removes iron more slowly to the extravascular space thereby allowing free Hb to be in the circulation for a longer period, causing more oxidative stress. On the other hand, Hp 1-1 protects the endothelium against oxidative damage, as it is the most biologically active.33 In this analysis, the Hp 2-1 phenotype was more common in both patients and controls (56.7% and 41.9%, respectively). Moreover, the profile of the Hp genotype was not different between the groups (p-value=0.70). This pattern agrees with most studies on the distribution of haplotype polymorphisms in SCD.32,34
Besides its antioxidant properties, Hp is an acute phase protein with immune-modulatory functions. It modulates the equilibrium between Th1 and Th2 lymphocytes by predominantly promoting Th1 cellular response. These cells are more effective in protecting against infections involving intracellular parasites and inhibiting the Th2 cytokines needed for humoral response.35 Moreover, the Hp-1-1-Hb complex significantly stimulates the secretion of more IL-6 than the Hp-2-2-Hb complex.14
There have been inconsistencies in the distribution of Hp genotypes in the SCA population. In 2011, Santos et al.32 reported that the Hp 2-2 phenotype in Brazilian SCA patients was less frequent than the Hp 1-1 phenotype and hypothesized that Hp 2-2 might be associated with worse prognosis because of lower antioxidant capacity and higher inflammatory response. In the current study, the Hp 1-1 phenotype was less common (16.7%), which agrees with the findings of Adekile & Haider in Kuwaiti patients with SCD in which only 4.9% had Hp 1-1 compared to 52.4% in those with the Hp 2-2 genotype.34 On the contrary, in the same study they reported Hp 2-2 as being less frequent among Nigerian patients with SCD. It was concluded that, in SCD individuals, the Hp genotypes appear to differ between ethnic groups and geographical areas, and are not necessarily linked to β-globin gene haplotypes in different populations.
Most studies on the influence of Hp polymorphisms on the clinical phenotypes of SCD have failed to establish any strong link between the two. For instance, no significant association was found between the Hp-2 allele and increased risk of severe vaso-occlusive crises among Kuwaiti Hb SS patients.34 This may imply that other factors mediate the influence of Hp. Of such, the scavenger receptor CD163 and heme oxygenase play active roles in the clearance of the Hb–Hp complex from the plasma, thereby preventing oxidative damage caused by heme.15 The level and activity of CD163, rather than Hp polymorphisms may therefore be related to SCD complications and severity.
Among the SCA patients in the present study, the genotypes did not appear to be related to the serum levels of IL-6 and IL-8; there was no relationship between the Hp genotypes and cytokine levels. This is in contrast to the findings of Guetta et al. who reported that the Hp-1 allele was associated with the secretion of significantly more IL-6 and IL-10 than the Hp-2 allele.14 They also demonstrated that the release of these cytokines was dependent on the binding of the Hb-Hp complex to the CD163 receptor and the activity of casein kinase II. However, the lack of association of Hp polymorphisms with the expression of pro-inflammatory cytokines may be due to the small number of patients in the present study.
Considering the important roles of cytokines in SCA pathophysiology, further investigations are necessary to identify factors that are responsible for increased secretion of plasma IL-6 and IL-8. Although the results of this study do not allow us to conclude that Hp genotypes on their own influence expression of IL-6 and 8 in SCA patients, this polymorphism when investigated with other genetic factors, may unravel the pathogenesis of elevated pro-inflammatory cytokines in SCA.
Conflicts of interestThe authors declare no conflicts of interest.
This study was funded by Fundação de Amparo à Pesquisa do Estado de São Paulo – FAPESP (grant number 04/04498-4) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – CAPES (grant number 054/2010).
Scholarships were provided by CAPES, CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico) and COLSAN (Associação Beneficente de Coleta de Sangue).

