




Gaucher disease is an inborn, autosomal recessive error of the metabolism which belongs to the group of lysosomal storage disorders.
ObjectiveThis work reports on the treatment of Gaucher disease in several members of the same family from the countryside of Maranhão.
MethodsThis was an observational, retrospective and prospective, descriptive case study about the efficacy of enzyme replacement therapy.
ResultsThe results showed that women were more affected (80% of patients) by the disease, age at diagnosis ranged from 24 to 33 years, the predominant ethnicity was mulatto (80%) and all cases were classified as type 1. The diagnosis of these patients was performed by measuring the levels of glucocerebrosidase and chitotriosidase enzymes and confirmed by genotyping. All patients suffering from Gaucher disease had low glucocerebrosidase levels. Before replacement therapy, hepatosplenomegaly was the most common clinical manifestation (100%) and osteopenia was seen in 80% of the cases. Regarding hematological manifestations, anemia and leukopenia were found in 40% of patients at diagnosis; however the hemoglobin and leukocyte levels were normalized after four years of therapy. Thrombocytopenia, observed in 20% of cases, was normalized after the second year of treatment.
ConclusionIn these cases, despite gaps in the treatment as the family resides in the rural region of the state, the patients with Gaucher disease showed satisfactory therapeutic response over time.
Gaucher disease (GD) was first described in 1882. It is a recessive autosomal lysosomal storage disorder which is caused by glucocerebrosidase deficiency that induces an accumulation of undigested glycolipids and leads to histological changes, especially evident in organs that are rich in elements of the monocytic-phagocyte immune system (spleen, liver and bone marrow).1,2 In more severe cases, this disease can affect the lungs, kidneys and central nervous system (CNS).3–5
GD is classified into three types according to the presence or absence of primary disease in the CNS: Type 1, the non-neuropathic form is the most common, Type 2, presents with serious impairment of CNS in childhood, and Type 3, with mild impairment of the CNS in adolescence or early adulthood.6
The most common type is also called the adult's non-neuropathic chronic form (90–99% of cases). It is prevalent among cases of Ashkenazi Jews, affecting about 1:450 of these individuals and 1:40,000 of the general population.7,8
Diagnosis is by biochemical tests that can demonstrate glucocerebrosidase deficiency with enzyme activity in leukocytes and cultured skin fibroblasts below 10% of the normal level. These values are determined by laboratory tests or by skin biopsy.4,9 A bone marrow biopsy is widely used to identify Gaucher cells, however, it is not pathognomonic and it can often lead to wrong diagnoses such as chronic myeloid leukemia, myeloblastic leukemia, Niemann-Pick disease, Hodgkin's disease and non-Hodgkin nodular lymphoma.7
GD is transmitted by autosomal recessive inheritance defined by mutations in the beta-glucosidase (GBA) gene that is responsible for encoding the glucocerebrosidase enzyme. This gene is located on the long arm of chromosome 1 (q21 region). The presence of two mutant alleles confirms diagnosis. The most prevalent mutations in this disease are N370S and L444P.10
The main clinical manifestations of GD include: hepatosplenomegaly, hematological disorders (anemia, thrombocytopenia and, more rarely, leukopenia), bone injuries and CNS impairment.11 Besides these symptoms, patients may also be affected by non-specific symptoms such as insomnia, headaches, chronic pain, paresthesia, depression and diffuse musculoskeletal pain. These patients do not have a good response to enzyme replacement therapy (ERT).
In addition, psychiatric symptoms, and more subtle, initial cognitive and motor changes may also appear.12,13
The treatment of most symptomatic patients is mainly supportive care with painkillers. Thus, the Brazilian Ministry of Health introduced ERT for GD in 1990 through the Special Drug Program, producing a protocol of correct therapeutic procedures.14,15
ERT has significantly changed treatment for Gaucher disease with reduced morbidity and improved quality of life and thus this is currently the treatment of choice; it is free through the Brazilian National Health Service.16
Herein we report the response to ERT of a group of patients with Gaucher disease in the same family from São Domingos do Maranhão. São Domingos do Maranhao is located 380km from the capital, São Luís, and according to the latest census, the population is 33,506 people.17,18 This study was approved in all respects by the Evaluation Committee for Research and Training (CAPPE) of HEMOMAR.
Case reportThis study included five patients with the diagnosis of GD of a family of 11 individuals from the municipality of São Domingos do Maranhão. All were treated by the Hematology and Hemotherapy Center of Maranhão (HEMOMAR) located in the state capital.
The instrument used for data collection included two forms to collect clinical, laboratory, radiological and socioeconomic data, besides the medical records from 2003 to 2010. Data on gender, race, age at diagnosis, occupation, classification according to the disease type, weight, height, glucocerebrosidase and chitotriosidase enzyme levels and type of genetic mutation were obtained from medical records.
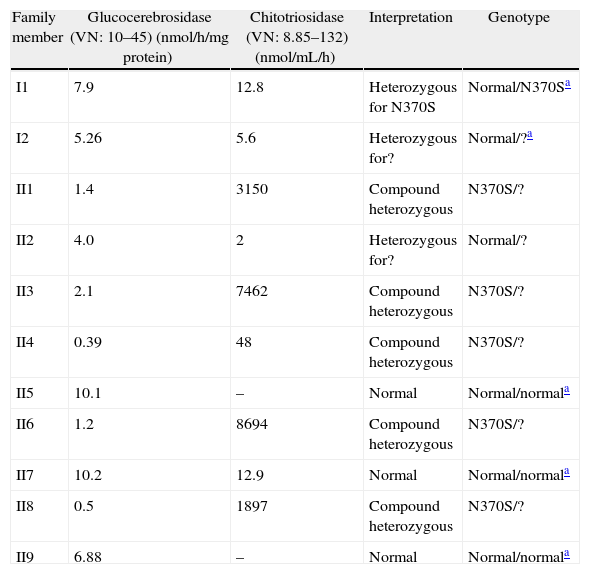
The family heredogram below shows three individuals heterozygous for GD (I1, I2 and II2), five compounded heterozygous for GD (II1, II3, II4, II6 and II8) and three normal individuals (II5, II7 and II9) (Fig. 1).

All patients had diagnostic confirmation of GD by measurement of glucocerebrosidase enzyme activity. The affected family members (heterozygotes) are: II1, II3, II4, II6, II8, all featuring the same genotype (N370S/?). Patient II2 is the only member of the second heterozygous generation for the unidentified allele (?), whose genotype is normal/? The other members of the second generation: II5, II7 and II9 are normal, with no significant changes in their enzyme measurements. Parents I1 and I2 are heterozygous for N370S and ? respectively. All patients affected by GD showed very high levels of chitotriosidase, except for Patient II4 (Table 1).
Glucocerebrosidase and chitotriosidase levels and the genotype of all family members.
| Family member | Glucocerebrosidase (VN: 10–45) (nmol/h/mg protein) | Chitotriosidase (VN: 8.85–132) (nmol/mL/h) | Interpretation | Genotype |
| I1 | 7.9 | 12.8 | Heterozygous for N370S | Normal/N370Sa |
| I2 | 5.26 | 5.6 | Heterozygous for? | Normal/?a |
| II1 | 1.4 | 3150 | Compound heterozygous | N370S/? |
| II2 | 4.0 | 2 | Heterozygous for? | Normal/? |
| II3 | 2.1 | 7462 | Compound heterozygous | N370S/? |
| II4 | 0.39 | 48 | Compound heterozygous | N370S/? |
| II5 | 10.1 | – | Normal | Normal/normala |
| II6 | 1.2 | 8694 | Compound heterozygous | N370S/? |
| II7 | 10.2 | 12.9 | Normal | Normal/normala |
| II8 | 0.5 | 1897 | Compound heterozygous | N370S/? |
| II9 | 6.88 | – | Normal | Normal/normala |
There is predominance of females (80%). Table 2 shows that all patients were diagnosed in adulthood with ages between 24 and 33 years and a mean of 28.4 years and are predominantly mulatto with only one Caucasian patient. Most patients are agricultural laborers (80%). All patients are type 1 or compose a characteristic non-neuropathic clinical picture.
Data on the determination of glucocerebrosidase and chitotriosidase levels were collected from medical records. Fig. 2 shows that only Patient #3 had a normal chitotriosidase level, while the others had high levels of this enzyme before starting ERT. After about seven years (in case of Patients #1–4) and three years (Patient #5) of ERT, all patients had chitotriosidase levels within the normal range.

The presence of anemia, leukopenia, thrombocytopenia, splenomegaly, hepatomegaly and skeletal changes were evaluated prior to and after starting ERT between 2003 and 2010.
Anemia was identified in 40% of patients at diagnosis (Fig. 3). However, after four years of ERT, there was normalization of hemoglobin levels in all patients with these levels remaining normal until 2010, except for Patient #4, who showed a decreased hemoglobin level in 2008.

At diagnosis, 40% of patients were leukopenic but after four years of ERT, no patient had leukopenia. In 2008, Patients #1 and #4 presented with leukopenia.
At diagnosis, 20% of patients had thrombocytopenia which was resolved after the second year of ERT with all patients having normal platelet counts. It is noteworthy that Patient #1 had no thrombocytopenia before ERT however the platelet count dropped in 2006.
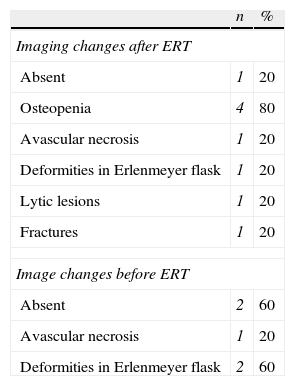
At diagnosis, there was bone impairment in 80% of patients. The most common osseous impairment prior the ERT was osteopenia, present in 80% of patients (Table 3). It seems that bone impairment is still present during ERT, with 20% of cases having Erlenmeyer flask deformity and 20% having avascular necrosis. Some patients had more than one lesion before the ERT but there were no imaging changes in 60% of cases after ERT (Table 3).
Bone impairment in patients with Gaucher disease before and after enzyme replacement therapy (ERT).
| n | % | |
| Imaging changes after ERT | ||
| Absent | 1 | 20 |
| Osteopenia | 4 | 80 |
| Avascular necrosis | 1 | 20 |
| Deformities in Erlenmeyer flask | 1 | 20 |
| Lytic lesions | 1 | 20 |
| Fractures | 1 | 20 |
| Image changes before ERT | ||
| Absent | 2 | 60 |
| Avascular necrosis | 1 | 20 |
| Deformities in Erlenmeyer flask | 2 | 60 |
Visceromegalies and hepatosplenomegaly were present in all of the patients before ERT however at the end of this study only 20% of patients had splenomegaly.
There was a significant gain in body mass of patients after starting ERT, except for Patient #5.
DiscussionGaucher disease is pan-ethnic, but with a high incidence in the population of Ashkenazi Jews, involving 1:450 live births compared to just 1:40.000 in the general population.7 Four alleles (N370S, L444P, 84GG, IVS2+1) account for most of the mutations that cause the disease, in particular in Ashkenazi Jews due to the tradition of marrying within the same ethnic group.7
This study focuses on a rare health problem of one family in Maranhão. According to reports, there is a very pertinent relationship to the origin of this family confirmed by the members who reported Jewish ancestry, but denied any relationship between the parents.7
The diagnosis of GD should be based on the measurement of the glucocerebrosidase enzyme in the presence of suggestive clinical manifestations of the disease. When this does not confirm diagnosis, other methods are used, such as high levels of chitotriosidase enzyme and family medical history, bone marrow biopsy, molecular analysis (genotyping), nonspecific imaging results and laboratory exams.19 In this family, four members (36.3%) had glucocerebrosidase activity suggestive of GD, four (36.3%) levels suggesting heterozygosity and two (18.1%) cases had normal enzyme activity. Family member II3, despite not having enzymatic values between 0 to 1.9nmol/h/mg protein, had confirmed GD diagnosis.
There is a strong relationship between these mutations and clinical manifestations of the disease. Thus, the genotype can help predict severity in patients. For instance, homozygosity for the N370S allele is associated with a less severe phenotype, although with wide clinical variety. Heterozygosity for N370S is protective against impairment of the SNC.19 The family members with GD were II1, II3, II4, II6, II8, all of whom had the N370S/? genotype (compound heterozygote). Family member II2 is female, heterozygous with the N/? genotype. The other individuals (II5, II7 and II9) are normal, with no significant changes in their enzyme levels.
An analysis of the probability of individuals having GD in a family whose parents are heterozygous shows that 25% of births are compound heterozygous (affected by GD), 50% heterozygous (one allele with a mutation of the GBA gene) and 25% are normal. In this case, of the total of nine children, five (55.5%) are compound heterozygotes for GD, thus showing the peculiarity of this case.
In a study conducted in Maranhão, 13 in a total of 14 patients with GD had documented genotyping. The N370S/? genotype was found in 42.2% of cases, whereas the presence of the N370S mutation was identified in more than two thirds of the patients studied.15 In a study from Santa Catarina State, the N370S allele was identified in 60% of cases, with homozygosity in only one case. The L444P mutant allele was present in heterozygosis with the N370S mutation in 20% of patients.7
GD is inherited in an autosomal pattern and can affect both genders. With a total of five patients affected by GD and all members of the same family, in this study there was predominance of female patients (80%). In Maranhão, females are predominant in 64.3% of cases.15 In one of the most important studies on GD, there was a slight predominance of females at 54%.20 In Brazil, out of the total of 551 patients registered and accompanied by the Gaucher Registry, 58% of patients are female. The situation is similar worldwide, with 53% females and 47% males of a total of 4764 patients enrolled in registries.21
This study reported that all patients are mulattoes except for one patient who is Caucasian. In a survey conducted in the state of São Paulo, a 62.2% rate of non-white patients was found for GD cases.22
ERT was introduced in 1991 and has been shown to reverse or ameliorate many of the visceral and hematological manifestations of GD. ERT also improves bone manifestations, including bone crises, bone pain, and appearance of new osteonecrosis in the joints and pathological fractures, and improves bone mineral density to varying degrees.23
After measuring enzyme levels and genotyping, all five patients affected by the disease in this family fall into the clinical classification of type 1 GD. Studies report that most patients are type 1 (94–96% type 1), less than 1% are type 2 and about 5% are type 3.8,24
Anemia has been reported in the literature in between 50%15 and 100%7 of cases. In the International Collaborative Gaucher Group (ICGG) report, anemia was present in 52% of cases before treatment with imiglucerase.21 In the study in Santa Catarina State, leukopenia was also present in 50% of cases.7 In an Italian study, hemoglobin, and leukocyte and platelet counts remained stable after ERT.25 In several studies thrombocytopenia was present: there was a significant percentage (50% of cases) in Maranhão.15 In one study, thrombocytopenia was found in 56.25% of 128 patients,26 partially disagreeing to data found in this study.
Hepatosplenomegaly was the most common clinical feature observed in patients with GD in the current study. In a study in Maranhão, hepatomegaly and splenomegaly affected 66.6% and 83.3% of patients, respectively.15 In another study, hepatosplenomegaly was the most common pathological finding at physical examination (80% of cases).7
There is a wide spectrum of skeletal complications related to GD, ranging from asymptomatic osteopenia to osteonecrosis, with secondary articular degeneration.27 A study conducted in the state of Santa Catarina, detected bone abnormalities in 75% of patients with the most common being osteopenia in 40% of cases, followed by avascular necrosis in 30% and Erlenmeyer flask deformity in 20%.7 In a study conducted in Spain, 40% of patients complained of diffuse bone pain.28 In a study carried out in Israel, after four years of ERT, there was a significant improvement in spleen and liver volumes, hemoglobin and platelet counts and Z-scores for lumbar spine and femur.23
Before starting the ERT, 80% of patients had a body mass index (BMI) between 18.5 and 24.9kg/m2. Only one of the patients had a BMI between 25 and 29.9kg/m2. However, in 2010, seven years after starting the ERT, 60% of patients entered the overweight threshold and 40% had normal BMIs. Among those considered normal, only one lost body mass (Patient #5 – from 27.6kg/m2 to 23.8kg/m2).
In conclusion, five members of this family had GD and were followed up at HEMOMAR. The age at diagnosis and classification of the GD type correlated to cases registered in the State of Maranhão with the prevalence of the N370S mutation. ERT improved hematological indexes, hepatosplenomegaly and the quality of life of these patients.
Conflicts of interestThe authors declare no conflicts of interest.



