



It has recently been estimated that each year, more than seven million babies worldwide are born with either a congenital abnormality or a genetic disease.1 Hemoglobinopathies are the commonest autosomal hereditary disorders and present a major public health problem in India. The overall prevalence of the β-thalassemia trait is 2.78% but this varies from 1.48 to 3.64% in different states of India2 compared to the carrier frequency in Brazil (1%).3 Recently we reported that the carrier frequency of the β-thalassemia gene with the IVS1-5 (G→C) mutation in western districts of Odisha, India is 3.75%.4,5 In India, the IVS1-5 (G→C) mutation is the most common β-thalassemia mutation. However, the IVS1-5 (G→C) mutation along with other mutations, including IVS1-1 (G→T), Cd41/42 (-TCTT), Cd 8/9 and a 619 base pair deletion, accounts for >90% of mutations causing β-thalassemia. In Brazil, the Cd 39 (C→T) mutation is the most prevalent cause of β-thalassemia followed by the IVSI-6 (T→C), and IVSI-110 (G→A) mutations.6 A map showing zonal distribution of β-thalassemia mutations in India is depicted in Figure 1.

The Veer Surendra Sai Medical College and Hospital, Burla, Odisha is a tertiary care referral hospital catering for western Odisha as well as eastern districts of Chhattisgarh state. Under a comprehensive sickle cell care program, we screen outpatient and hospitalized cases in the Institute for Sickle Cell Disease and other hemoglobinopathies at the Sickle Cell Clinic and Molecular Biology Laboratory.
Compound heterozygotes for β-thalassemia and structural hemoglobin (Hb) variants usually present with a severe form of the disease. Here, we report a case, a compound heterozygote for the β-thalassemia mutation IVS1-5 (G→C) and Indian deletion-inversion Gγ(Aγδβ)0-thalassemia (HbVar ID-1038) from the district of Bargarh in the state of Odisha, India. The molecular structure of the Indian deletion-inversion [Gγ(Aγδβ)0-thal] is caused by a major rearrangement including an inversion of the sequence between the 3′ end of the Aγ gene and the IVS-II region of the β-globin gene, followed by two deletions (total 8.5kb) of the flanking DNA sequence. This is the first case report of such a case from eastern India.
Case reportAn 18-year-old female patient (height 147cm and weight 40kg) belonging to the ‘Chasa’ caste was admitted to the Department of Medicine of Veer Surendra Sai Medical College and Hospital, Burla, Odisha, India with severe anemia, but with no history of any other medical complaints. Her Hb level was 2g/dL at the time of admission to the hospital. After a transfusion of three blood units, the Hb level reached 7.8g/dL. The erythrocyte sedimentation rate was 12mm/h. Other parameters of blood indices after the blood transfusion were white blood cell count (WBC): 10.9×109 cells/L; red blood cells (RBC): 3.35×106/μL; hematocrit (HCT): 23.9%; mean corpuscular volume (MCV): 71.7fL; mean cell hemoglobin (MCH): 20.6pg; mean cell hemoglobin concentration (MCHC): 29.0g/dL; and platelets (PLT): 149×109/L. Peripheral blood smear examination showed microcytic, hypochromic anemia, marked anisopoikilocytosis, many microcytes, few macrocytes and fragmented RBCs. Staining by the Kleihauer–Betke method demonstrated pancellular distribution of fetal hemoglobin (Hb F). Ultrasonography examination revealed hepatomegaly (16.5cm) and splenomegaly (14.5cm). An X-ray of chest (P-A view) and X-ray of both hip joints (A-P view) showed no abnormalities.
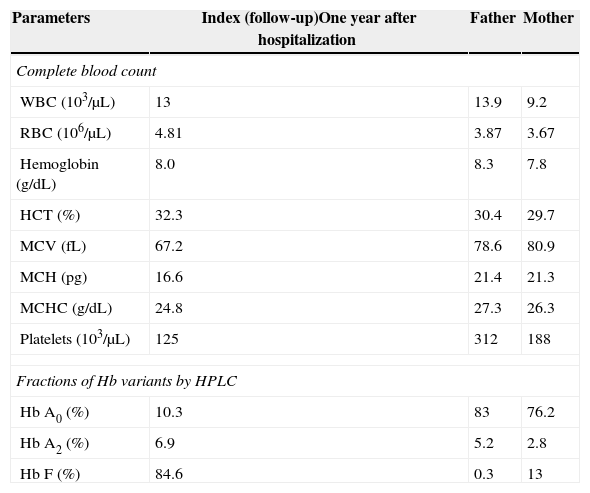
Automated high-performance liquid chromatography using the β-Thalassemia Short Program on Bio-Rad Variant-II system showed various fractions of Hb with a raised level of Hb F (84.6%) and an Hb A2 concentration of 6.9% (Figure 2). Parents’ studies revealed that her mother had a high Hb F level (13%) whereas her father had high Hb A2 level (5.2%). All hematological parameters of the case and her parents are shown in Table 1. Screening for the common molecular determinants of raised Hb F, the Indian deletion-inversion Gγ(Aγδβ)0-thalassemia and hereditary persistence of fetal Hb (HPFH) was performed by gap-polymerase chain reaction (PCR) using primers and protocol as described previously.7 Common Indian β-thalassemia mutations were confirmed by multiplex amplification refractory mutation system (ARMS)-PCR using a previously described protocol.4 Moreover, alpha globin gene deletions (α−3.7 and α−4.2) were investigated by gap-PCR.8 The DNA study showed father as heterozygous for the IVS1-5(G→C) mutation and mother as a Gγ(Aγδβ)0-thalassemia carrier. The gel picture for Indian deletion-inversion Gγ(Aγδβ)0-thalassemia is shown in Figure 3. Alpha-thalassemia was not observed in any of them.
 mutation and Indian deletion-inversion Gγ(Aγδβ)0 thalassemia (chromatogram one year after hospitalization at follow-up).")
Hematological investigations in the case of compound heterozygous state for β-thalassemia with IVS1-5 (G→C) mutation and Indian deletion-Inversion Gγ(Aγδβ)0 thalassemia with her parents.
| Parameters | Index (follow-up)One year after hospitalization | Father | Mother |
|---|---|---|---|
| Complete blood count | |||
| WBC (103/μL) | 13 | 13.9 | 9.2 |
| RBC (106/μL) | 4.81 | 3.87 | 3.67 |
| Hemoglobin (g/dL) | 8.0 | 8.3 | 7.8 |
| HCT (%) | 32.3 | 30.4 | 29.7 |
| MCV (fL) | 67.2 | 78.6 | 80.9 |
| MCH (pg) | 16.6 | 21.4 | 21.3 |
| MCHC (g/dL) | 24.8 | 27.3 | 26.3 |
| Platelets (103/μL) | 125 | 312 | 188 |
| Fractions of Hb variants by HPLC | |||
| Hb A0 (%) | 10.3 | 83 | 76.2 |
| Hb A2 (%) | 6.9 | 5.2 | 2.8 |
| Hb F (%) | 84.6 | 0.3 | 13 |
WBC: white blood cells; RBC: red blood cells; HCT: hematocrit; MCV: mean cell volume; MCH: mean cell hemoglobin; MCHC: mean cell hemoglobin concentration; HPLC: high-performance liquid chromatography.
![Agarose gel electrophoresis for the detection of β-thalassemia with IVS1-5 (G→C) mutation (ARMS PCR) and Indian deletion-inversion Gγ(Aγδβ)0 thalassemia (GAP-PCR). *I: index; M: mother; F: father; L: ladder. Detection of β-thalassemia with IVS1-5 (G→C) mutation by ARMS PCR (Line A1, Line A4 and Line A6 for normal β-globin gene, and Line A2, Line A5 and Line A7 for IVS1-5 [G→C] mutation). Detection of Indian deletion-inversion Gγ(Aγδβ)0 thalassemia by GAP-PCR (Line B1, Line B2 and Line B3 for Breakpoint-A, and Line B4, Line B5 and Line B6 for Breakpoint-B).](https://static.elsevier.es/multimedia/15168484/0000003700000003/v1_201506010548/S1516848415000584/v1_201506010548/en/main.assets/gr3.jpeg?xkr=ue/ImdikoIMrsJoerZ+w92xcqUtYYCc6yiYRjTD2AR1aFJOKb7qLd+WBf18Ptk/O8dVG5SMqADZji5skBxdmL5qkYw7MDAvd7AU2+H4Vkulsu6/ST6T8eZyS9nePvuO89xxKaEEmDoj6skIcP4DVEyBGY2GFZOuSxTRTmof5usM92Z9ki23jyXXJ8a2Y07ThxrkVW2kcJ5tcgBc2/IG4PX1ydmSPvjoVHm/1rq64pktkapBp+xxYNMcP4JnsMAxt5Fna77HOs0BvQ9cNPZO/Qa612R+HAQ6MO++ZIwQCMcf8E/y1AqnNOE7mmrkIAocS "Agarose gel electrophoresis for the detection of β-thalassemia with IVS1-5 (G→C) mutation (ARMS PCR) and Indian deletion-inversion Gγ(Aγδβ)0 thalassemia (GAP-PCR). *I: index; M: mother; F: father; L: ladder. Detection of β-thalassemia with IVS1-5 (G→C) mutation by ARMS PCR (Line A1, Line A4 and Line A6 for normal β-globin gene, and Line A2, Line A5 and Line A7 for IVS1-5 [G→C] mutation). Detection of Indian deletion-inversion Gγ(Aγδβ)0 thalassemia by GAP-PCR (Line B1, Line B2 and Line B3 for Breakpoint-A, and Line B4, Line B5 and Line B6 for Breakpoint-B).")
Agarose gel electrophoresis for the detection of β-thalassemia with IVS1-5 (G→C) mutation (ARMS PCR) and Indian deletion-inversion Gγ(Aγδβ)0 thalassemia (GAP-PCR). *I: index; M: mother; F: father; L: ladder. Detection of β-thalassemia with IVS1-5 (G→C) mutation by ARMS PCR (Line A1, Line A4 and Line A6 for normal β-globin gene, and Line A2, Line A5 and Line A7 for IVS1-5 [G→C] mutation). Detection of Indian deletion-inversion Gγ(Aγδβ)0 thalassemia by GAP-PCR (Line B1, Line B2 and Line B3 for Breakpoint-A, and Line B4, Line B5 and Line B6 for Breakpoint-B).
Written informed consent was obtained from the patient along with her parents and the study was approved by Institutional Ethics Committee of Veer Surendra Sai Medical College and Hospital, Burla, Odisha, India.
Discussionβ+-Thalassemia is the second most common hemoglobinopathy in our clinic's population. We have reported earlier the clinical and molecular characteristics of Gγ(Aγδβ)0-thalassemia in heterozygous as well as in compound heterozygous states with Hb S. In Hb S/Gγ(Aγδβ)0-thalassemia cases, the patients had repeated painful crises along with histories of blood transfusions.9 Indian deletion-inversion Gγ(Aγδβ)0-thalassemia in the heterozygous form has been reported in western India in the state of Maharashtra.10 Recently, Pandey et al. described seven cases with Indian deletion-inversion Gγ(Aγδβ)0-thalassemia, of which three cases were co-inherited with β-thalassemia with the IVS1-5(G→C) mutation.11 These three cases had low values of both Hb A2 (3.2%, 2.8%, and 2.7%) and Hb F (21.3%, 10.7% and 20.3%) compared to the 6.9% and 84.6% of Hb A2 and Hb F, respectively in the current case. Their three cases were transfusion dependent and showed moderate anemia. Our case is the first case of compound heterozygote state of β-thalassemia with IVS1-5(G→C) mutation and Gγ(Aγδβ)0-thalassemia with mild phenotype from eastern India. In this patient, there was no other probable cause to explain the acute episode of anemia in history or investigations. As parvovirus B19 is not investigated in our institution, this may well be a cause of the anemia in this patient. The milder clinical presentation is likely due to the association of a milder β+-thalassemia allele with high Hb F level (84.6%). Our observation of homogeneous F cell distribution and an elevated Hb F level in peripheral blood, is in agreement to the report of Waye et al. in an African-American with compound heterozygosity of a Black form of (Aγδβ)0-thalassemia and the −29 (A→G) β+-thalassemia mutation.12 We only investigated two types of α-thalassemia (α−3.7 and α−4.2), the most prevalent in India. Another form of α-thalassemia may be a factor for milder clinical presentation of the patient.
The Indian subcontinent has a heterogeneous population with different hemoglobinopathies. These Hb disorders should be included in the prenatal diagnosis of patients with severe or mild thalassemia. The characterization of these hemoglobinopathies will facilitate a prevention and control program of hemoglobinopathies including thalassemia in this region.
Conflicts of interestThe authors declare no conflicts of interest
This study was supported by research funding from Indian Council of Medical Research (ICMR), New Delhi, Department of Science and Technology (DST), New Delhi, and National Health Mission (NHM), Odisha.



