



Iron overload (IO) is a complex condition in which clinical, behavioral and genetic factors contribute to the phenotype. In multiethnic and non-Caucasian populations, mutations in HFE gene alone cannot explain IO in most of the cases, and additional genetic and environmental factors must be investigated. Bone Morphogenetic Proteins (BMPs) play a central role in iron homeostasis by modulating HAMP transcription through the signaling pathway that includes SMAD and HJV. In this study, we aimed to explore the clinical relevance of BMP6 mutations in a cohort of Brazilian patients with IO.
Methods41 patients with IO were evaluated. Blood samples were collected to analyze BMP6 mutations through New Sequence Generations (NGS). Frequency of variants and mutations were analyzed and correlated with clinical and environmental characteristics.
ResultsWe identified BMP6 mutations in three patients with IO. The p.Arg257His mutation was identified in two patients and the p.Leu71Val mutation was identified in one patient. Two of these patients had additional risk factors for IO (HFE mutations and diabetes mellitus).
ConclusionBMP6 mutations, when combined to other genetic and clinical risk factors, may contribute to IO. Functional studies and THE evaluation of large cohorts are necessary to fully address BMP6 role in IO.
Hemochromatosis is a disease characterized by iron overload (IO) caused by a genetic condition in which hepcidin-ferroportin system is affected and the plasma levels of hepcidin is low. The most common cause of hereditary hemochromatosis in Caucasians is a C282Y homozygosis mutation in HFE gene. Mutations in HJV, TFR2, SLC40A1, and HAMP genes have been associated to hemochromatosis, although they are less frequent.1 Nonetheless, people can develop hemochromatosis owing to a variety of conditions that include other gene mutations. Very probably co-morbidities and behavioral factors play an important role in the phenotype of the disease.2,3
Bone Morphogenetic Proteins (BMPs) play a central role in iron homeostasis.4 BMP6 is produced by the liver cells according to iron stores and, through the signaling pathway that includes SMAD and HJV, can modulate HAMP transcription and consequently hepcidin levels.1 Daher et al. described three heterozygous missense mutations in BMP6 (p.Pro95Ser, p.Leu96Pro, and p.Gln113Gl) in six patients with iron overload in whom hepcidin levels were low.5 Piubelli et al. described the BMP6 p.Arg257His mutation, which is probably pathogenic due to its location in the protein pro-peptide domain, that is necessary to BMP6 processing and secretion.1 The p.Val394Met BMP6 mutation was described by Alvarenga et al. and its clinical significance is still uncertain.6 In this study, we aimed to further explore BMP6 mutations in terms of frequency and clinical relevance in a cohort of Brazilian patients with IO and to correlate with additional genetic and clinical features.
Patients and methodsWe prospectively evaluated 41 patients (5 females, 36 males, median age at diagnosis 50 years (31–86 yo) with the diagnosis of IO followed at the Hematology and Hemotherapy Center of the University of Campinas, Brazil. IO was defined by hyperferritinemia (>200 ug in females; >300 ug in males) with transferrin saturation>45 % or evidence of IO on T2* magnetic resonance imaging (MRI). Clinical and laboratory characteristics from patients were collected by medical records evaluation.
Peripheral blood samples were obtained and genomic DNA was extracted from leukocytes using the phenol-chloroform technique. DNA was quantified using a Qubit Fluorometer (Thermo Fisher Scientific, Waltham, MA). A total of 50–500 ng of genomic DNA was fragmented to 140–230 bp by sonication, as measured using a Bioanalyzer 2100 instrument (Agilent Technologies, Santa Clara, CA). For the exome capture platform, genomic DNA libraries were constructed with SureSelect Target Enrichment Kit (Agilent Technologies). Enriched libraries were PCR amplified, pooled, and sequenced on a HiSeq 2500 (Illumina, San Diego, CA) using paired-end 2 × 150–bp configuration with a minimum read depth of the target region was above 100X.
The sequences obtained were aligned to the human reference genome (GRCh37/hg19) using the BWA program7 and analyzed using Picard (https://broadinstitute.github.io/picard/) and GATK48 to identify variants relevant to the clinical specific indication. Reports were issued on quality control, alignment of reads against the reference human genome GRCh37/hg19 and removal of duplicates. Quality control measures included a minimum average coverage of 150 reads at more than 90 % of positions within a target gene with coverage greater than 100X. The identified variants were recorded in a VCF file (Variant Call Format) and annotated with the wANNOVAR and InterVar algorithms.9,10 Filtering was performed with VarAFT11 which included the following criteria: non-synonymous variants, all coding and flanking regions adjacent to exons, splicing sites, 5′-UTR (untranslated region). Reading depth >30 and VAF (variant allele frequency) < 50 %. The variants selected for iron metabolism disorder were considered to be those with 0.1 % maximum allele frequency in the population as reported in 1000 genomes12 gnomAD database,13 Exome Variant Server (EVS, http: //evs.gs.washington.edu/EVS/) and ABraOM (Arquivo Brasileiro Online de Mutações)14 were eligible, considering the clinical condition of the patient. Variants were assessed by mutation prediction and conservation programs including SIFT,15 Polyphen-2,16 PROVEAN,17 MutationTaster2.18 Then, the relevant variants were submitted to clinical interpretation according to the guidelines established by the ACMG19 and those variants of unknown significance were manually reviewed.
This research was approved by the Institutional Review Board (number 41684915800005404) and has been carried out in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki). All participants signed the written informed consent.
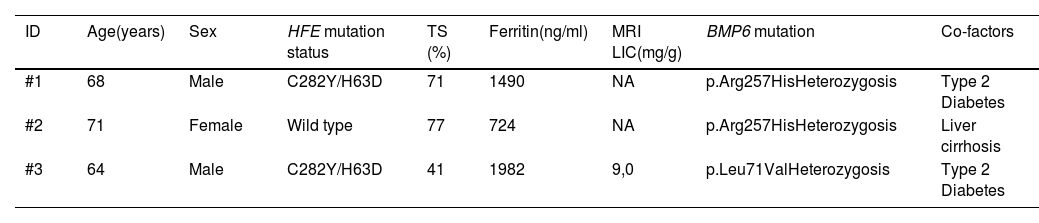
ResultsWe identified BMP6 mutations in three out of 41 patients with IO (7 %). The p.Arg257His was identified in two patients and the p.Leu71Val mutation was identified in one patient. Among the patients with p.Arg257His mutation, the first was a 68-year-old male who carried a compound heterozygous mutation in HFE gene (H63D/C282Y) and presented diabetes mellitus. The second patient was a 71-year-old female in whom no HFE mutations or environmental risk factors, were identified. She presented a severe liver cirrhosis with a grade 4 hepatic siderosis on liver biopsy without a clear etiology. The third patient was a 64-year-old male who also carried a compound heterozygous mutation in HFE gene (H63D/C282Y) and diabetes mellitus. He presented a moderate IO on the MRI. The p.Leu71Val mutation has not been previously described in the literature. The structure of the protein was analyzed using the Phyre2 software and the in silico analyses demonstrated that this seems not to cause a severe distortion of true molecule (Figure 1). Clinical and laboratory characteristics of the patients are shown in Table 1.
 and p.Leu71Val mutation (B).")
Clinical and laboratory characteristics of the patients with IO and BMP6 mutations.
MRI LIC, magnetic resonance image – liver iron concentration; TS, transferrin saturation; NA, not available.
Iron overload disorders have been described as a complex condition in which a variety of factors can contribute for its severity. The diagnosis of IO is associated not only with genetic, but also with clinical and behavioral factors.3 In this study, we identified two different mutations in IO patients: a known p.Arg257His mutation, which was a previously described as a pathogenic variant, and the p.Leu71Val mutation, that was not yet described.
Mutations in BMP6 gene have been increasingly described as an important issue in the context of IO.4 Borgel et al. showed that mutations in the pro-peptide region of BMP6 gene may be associated with defective protein secretion and, consequently, deficient hepcidin secretion.20 The p.Arg257His mutation was described in two previous studies that evaluated IO. According to these studies, this mutation appears to be pathogenic and related with the clinical phenotype of IO.1,6 Here we described two patients with the p.Arg257His mutation. The clinical phenotype of patients are in accordance with the characteristics described in the studies by Piubelli et al.1 and Alvarenga et al.,6 in which the patients who carried the p.Arg257His mutation presented moderate to severe IO, corroborating the possibility that this mutation is related to the IO phenotype.
In the study described here, the p.Leu71Val mutation, located in the pro-peptide domain of BMP6 gene, was identified in a male patient with the compound heterozygous HFE mutation C282Y/H63D. Previous data have shown that the C282Y/H63D HFE mutation itself has no significant clinical consequences and cannot explain IO alone.21,22 To further explore whether this new mutation could be associated with the IO phenotype, we analyzed the protein structure to predict modifications in the molecule and consequently in its function, but no structural mutation was identified. Daher et al. described the p.Leu96Pro mutation associated with IO, which also occurs in the pro-peptide domain, a region important for protein processing and secretion.5 The mutations are located in the same region, which may indicate the possibility of the association between the p.Leu71Val mutation and IO.
ConclusionIO is a condition which can be explained by genetic, clinical and behaviors factors. Among genetic features, a mixed pool of mutations with a variety of penetrance could explain different IO grades of severity. Further studies are necessary to identify not only the prevalence of BMP6 mutations in larger cohorts, but also its influence on the phenotype of the patients with IO. Given the significant ethnic heterogeneity of Brazilian population, the data presented is important to recommend that mutations other than HFE should be analyzed in patients with IO of unknown etiology.
This work was supported by the São Paulo Research Foundation (FAPESP) and the National Council for Scientific and Technological Development (CNPq) – Brazil.


