



Hemochromatosis is currently characterized by the iron overload caused by hepcidin deficiency. Large advances in the knowledge on the hemochromatosis pathophysiology have occurred due to a better understanding of the protein of the iron metabolism, the genetic basis of hemochromatosis and of other iron overload diseases or conditions which can lead to this phenotype. In the present review, the main aims are to show updates on hemochromatosis and to report a practical set of therapeutic recommendations for the human factors engineering protein (HFE) hemochromatosis for the p.Cys282Tyr (C282Y/C282Y) homozygous genotype, elaborated by the Haemochromatosis International Taskforce.
Hemochromatosis is currently characterized by the iron overload caused by a hepcidin deficiency. This hormone is a key iron modulator in the control of the ferroportin, an iron exporter protein in the cells. In hemochromatosis, plasmatic concentration of hepcidin is low in most cases. Thus, ferroportin activity is high in the enterocytes and, consequently, leads to high uptake and iron overload. But in rare cases, as in the ferroportin deficiency, there is a resistance to hepcidin. Large advances in the knowledge on the hemochromatosis pathophysiology occurred due to a better understanding of the protein in the iron metabolism, the genetic basis of hemochromatosis and of other iron overload diseases or conditions which can lead to this phenotype.1-3
In 2018, a Taskforce of the Hemochromatosis International (HI) published a practical set of recommendations on the therapeutic aspects of HFE hemochromatosis for the p.Cys282Tyr (C282Y/C282Y) homozygous genotype, based on published scientific studies and guidelines.4 In this scenario, physicians have the tools that allow them to make a differential diagnosis and to adequately manage the hemochromatosis patients.
The main aims of this study are to show updates on hemochromatosis and to report some aspects of the published therapeutic recommendations.
Diagnosis: clinical aspects, biochemical, genetic and imaging testsClinical aspectsPatients can be asymptomatic for decades and subsequently show the manifestation of symptoms for HFE hemochromatosis at approximately 40 years of age in men and 50 years of age in women. Patients with HFE hemochromatosis usually present non-specific symptoms, such as chronic fatigue, abdominal pain, skin hyperpigmentation, arthropathies, diabetes mellitus, hepatomegaly and cirrhosis (when the serum ferritin is > 1,000µg/L). A current study on the quality of life (QoL) showed that patients with the HFE p.Cys282Tyr homozygous genotype had a worse QoL, measured by the short form health survey (SF-36) scale, compared to patients with other genotypes.6
Biochemical testsLaboratory tests routinely used to investigate biochemical evidence of iron overload are the serum ferritin (SF) and transferrin saturation (TS). Box 1 shows values for SF and TS indicated by therapeutic recommendations from the HI taskforce.
The SF is a highly sensitive test for measuring iron overload. However, ferritin is an acute-phase protein and, consequently, elevated SF values are very frequently associated with other causes, such as general inflammations, metabolic syndrome, alcohol consumption and liver damages.2,3,7 It is pivotal to identify the cause of high SF values. SF values > 300µg/L in males and postmenopausal females and > 200 µg/L in premenopausal females were indicated in the therapeutic recommendations from the HI taskforce, in the diagnosis scenario.4
The TS is the ratio between serum iron and total iron-binding capacity (TIBC), expressed as a percentage. This is performed with patient fasting and it can be a helpful biomarker of iron overload when the values are ≥ 45% and it corresponds to the basic and earliest biochemical abnormality in the hemochromatosis. The interpretation of the SF and TS values needs to be performed for each individual, taking into account age, gender and co-morbidities, as well as local reference values.8,9
Genetic testsThe HFE hemochromatosis is an autosomal recessive disease and the most frequent genotype associated with the phenotype is the p.Cys282Tyr homozygosity (C282Y/C282Y). An important piece of information is that the penetrance of the HFE hemochromatosis is incomplete, i.e., from a low to moderate frequency of individual carrying homozygous genotype (C282Y/C282Y) will have clinical symptoms.
Patients with iron overload carrying the HFE p.Cys282Tyr/p.His63Asp compound heterozygous or p.His63Asp homozygous genotypes are frequently identified in some cohort studies on hemochromatosis, especially in countries with admixed populations. However, a study concerning best practices for the molecular genetic diagnosis of hemochromatosis concluded, according to the level and strength of the evidence, that both genotypes had the following interpretations: at low risk for development of significant iron overload (may be at risk of developing mild to moderate iron overload in association with comorbid factors), or at no increased risk of developing HFE hemochromatosis.10
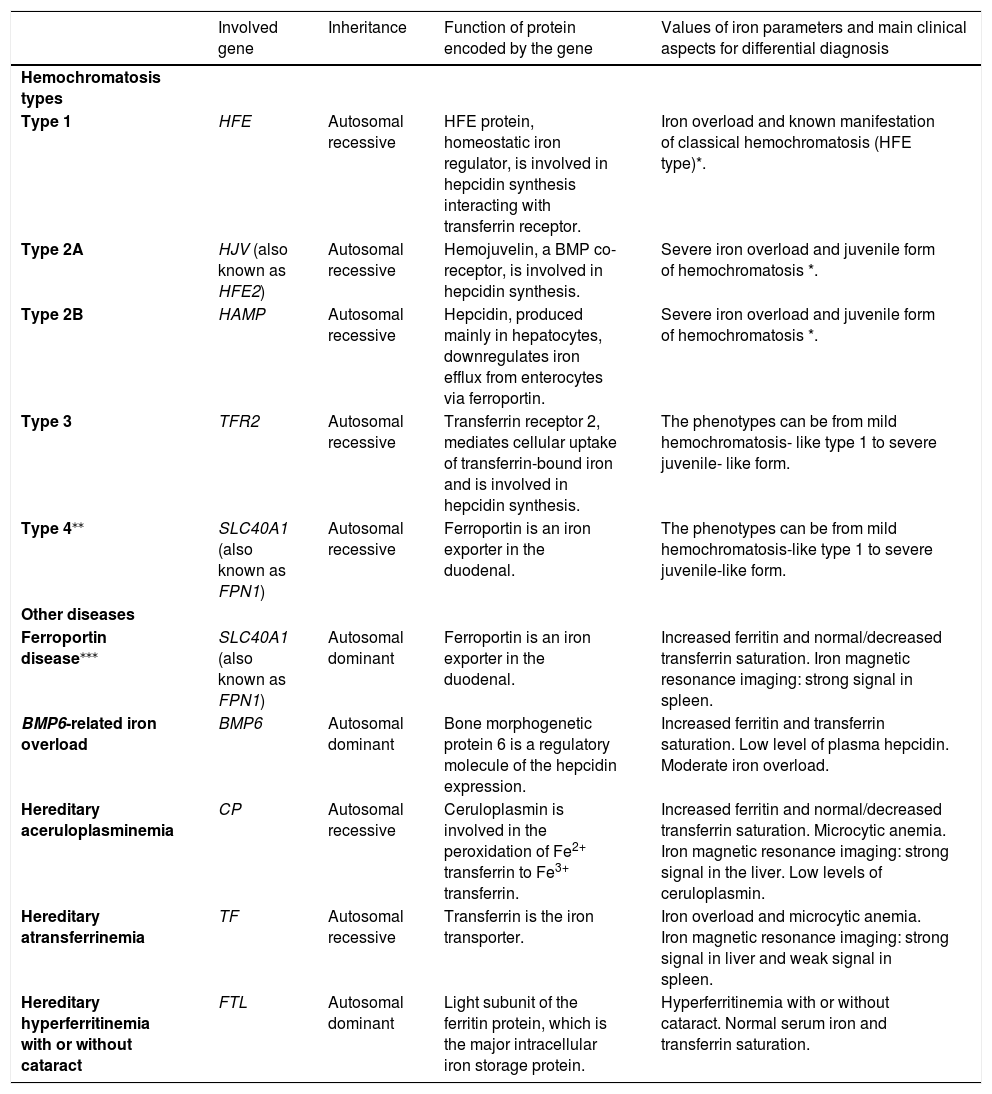
Patients with hemochromatosis not linked with HFE (non-HFE hemochromatosis) are usually younger patients aged 20 to 30 years old, presenting symptoms, such as heart failure, diabetes and hypogonadism.3,5 Non-HFE hemochromatosis includes very rare forms, associated with mutations in the HJV, HAMP, TFR2 and SLC40A1 genes. Genetic investigation for the non-HFE hemochromatosis will be needed when the result for the p.Cys282Tyr homozygosity is negative, mainly in the case of a younger patient with manifestations of the juvenile form and severe iron overload.11-14Table 1 shows data on the hemochromatosis types and some other genetic diseases that present abnormal values of iron parameters and thus, participate in the context of the differential diagnosis for hemochromatosis.
Data on hemochromatosis types and some genetic diseases that present abnormal values of iron parameters, suggesting hemochromatosis.
| Involved gene | Inheritance | Function of protein encoded by the gene | Values of iron parameters and main clinical aspects for differential diagnosis | |
|---|---|---|---|---|
| Hemochromatosis types | ||||
| Type 1 | HFE | Autosomal recessive | HFE protein, homeostatic iron regulator, is involved in hepcidin synthesis interacting with transferrin receptor. | Iron overload and known manifestation of classical hemochromatosis (HFE type)*. |
| Type 2A | HJV (also known as HFE2) | Autosomal recessive | Hemojuvelin, a BMP co-receptor, is involved in hepcidin synthesis. | Severe iron overload and juvenile form of hemochromatosis *. |
| Type 2B | HAMP | Autosomal recessive | Hepcidin, produced mainly in hepatocytes, downregulates iron efflux from enterocytes via ferroportin. | Severe iron overload and juvenile form of hemochromatosis *. |
| Type 3 | TFR2 | Autosomal recessive | Transferrin receptor 2, mediates cellular uptake of transferrin-bound iron and is involved in hepcidin synthesis. | The phenotypes can be from mild hemochromatosis- like type 1 to severe juvenile- like form. |
| Type 4⁎⁎ | SLC40A1 (also known as FPN1) | Autosomal recessive | Ferroportin is an iron exporter in the duodenal. | The phenotypes can be from mild hemochromatosis-like type 1 to severe juvenile-like form. |
| Other diseases | ||||
| Ferroportin disease⁎⁎⁎ | SLC40A1 (also known as FPN1) | Autosomal dominant | Ferroportin is an iron exporter in the duodenal. | Increased ferritin and normal/decreased transferrin saturation. Iron magnetic resonance imaging: strong signal in spleen. |
| BMP6-related iron overload | BMP6 | Autosomal dominant | Bone morphogenetic protein 6 is a regulatory molecule of the hepcidin expression. | Increased ferritin and transferrin saturation. Low level of plasma hepcidin. Moderate iron overload. |
| Hereditary aceruloplasminemia | CP | Autosomal recessive | Ceruloplasmin is involved in the peroxidation of Fe2+ transferrin to Fe3+ transferrin. | Increased ferritin and normal/decreased transferrin saturation. Microcytic anemia. Iron magnetic resonance imaging: strong signal in the liver. Low levels of ceruloplasmin. |
| Hereditary atransferrinemia | TF | Autosomal recessive | Transferrin is the iron transporter. | Iron overload and microcytic anemia. Iron magnetic resonance imaging: strong signal in liver and weak signal in spleen. |
| Hereditary hyperferritinemia with or without cataract | FTL | Autosomal dominant | Light subunit of the ferritin protein, which is the major intracellular iron storage protein. | Hyperferritinemia with or without cataract. Normal serum iron and transferrin saturation. |
Serum ferritin and transferrin saturation values indicated by therapeutic recommendations from the HI taskforce.
| Section*“Whom to treat and when to start” |
| Patients with the HFE p.Cys282Tyr (C282Y/C282Y) homozygous genotype and biochemical evidence of iron overload, i.e., increased serum ferritin (> 300µg/L in males and postmenopausal females and > 200 µg/L in premenopausal females) and increased fasting transferrin saturation (≥ 45%). |
Magnetic resonance imaging (MRI), when available, may be useful to measure parenchymal iron overload, mainly in the liver, but additionally in other organs, such as the heart, pancreas, spleen and pituitary. Iron overload in other organs besides the liver can be a suggestion of the juvenile form. In addition, the MRI can be used for the differential diagnosis of hemochromatosis with other genetic diseases (Table 1). Regarding the hemochromatosis evaluation, the MRI and laboratorial tests have been preferred over the liver biopsy, except in rare cases.2,3,19
TreatmentPhlebotomy (venesection therapy) is the standard treatment for patients with hemochromatosis and it is very effective to prevent hemochromatosis damages, safe and has a low cost. Thus, the early diagnosis and initiation of the phlebotomies are important actions to prevent tissue and cell damages due to reactive oxygen species from the iron overload. One phlebotomy of 450mL removes approximately 225mg of iron from the body.
Therapeutic recommendations from the HI Taskforce divided the treatment into two phases: initial phase (or induction) and maintenance phase (Box 2). The authors considered that a high percentage of the recommendations is not shared among the different available guidelines for hemochromatosis and therefore, has proposed objective recommendations which may be used for managing hemochromatosis patients throughout the world.4,20
Treatment indicated by therapeutic recommendations from the HI taskforce.
| Section*“How to treat” | |
| Initial or induction phase | Maintenance phase |
| Frequency | Frequency |
| A phlebotomy schedule on the order of 400 - 500mL, considering body weight, weekly or every two weeks has been proposed. | One phlebotomy every 1 to 4 months, depending on the patient's iron status.14,21 |
| Objective and tolerance | Objective and tolerance |
| Usually, to reach serum ferritin of 50µg/L.Serum ferritin should be checked once a month until the values reach the upper normal limits and every two weeks thereafter until the final goal of serum ferritin is reached.Hemoglobin levels should not decrease below 11 g/dL. | Usually, to maintain ferritin levels around 50µg/L.Hemoglobin levels should not be < 11 g/dL. |
There are several peculiarities of each hemochromatosis patient and considerations about treatment which must be pondered. Frequencies of the phlebotomies indicated in Box 2 for both phases (initial and maintenance) are significantly different among phases and within the phases. Thus, the phlebotomy frequency should be individualized to the clinical data for each moment of the management of the individualized hemochromatosis patient. Some elderly patients with comorbidities and/or poor vein conditions do not tolerate the “standard phlebotomy regimen”; in these patients, a modified phlebotomy regimen may be required in the induction phase, in which less than 400 to 500 mL blood is drawn and/or the intervals between phlebotomies may be extended and a higher ferritin level of 200 to 400 µg/L may be accepted to avoid anemia and the discomfort due to the phlebotomies.4
Patients who have had iron overload should never stop checking his or her iron parameters and should be followed lifelong on an out-patient basis.4
Iron chelation therapy is not indicated for classical hemochromatosis. However, in rare cases, iron chelators are an adjuvant treatment (or alternative), such as in severe iron overload without efficacy with phlebotomies, poor vein conditions and severe non-HFE hemochromatosis (mainly juvenile form).24,25 Erythrocytapheresis have been used to treat hemochromatosis patients, but is more expensive and less available than phlebotomy. Regarding diet, the hemochromatosis patient can have a normal healthy varied diet. Vitamin C, high alcohol consumption and foods with iron fortification should be avoided.2,4
Two current studies showed the importance of the adequate hemochromatosis treatment and of the early intervention to prevent morbidity caused by the hemochromatosis related to the HFE p.Cys282Tyr homozygosity. Ong and colleagues, enrolling patients with the p.Cys282Tyr homozygous genotype and with a moderate serum ferritin level (300 - 1,000µg/L), performed a randomized controlled trial by dividing the cohort into two groups: iron reduction by erythrocytapheresis (treatment) or sham treatment by plasmapheresis (control). They identified an improvement in the modified fatigue impact scale (MFIS) score in the treatment group, compared to the control. Pilling and colleagues, in a large cohort study at the UK biobank including 2,890 patients with the p.Cys282Tyr homozygous genotype, concluded that hemochromatosis was associated with significant prevalent and incident clinically diagnosed morbidity (liver disease, rheumatoid arthritis, osteoarthritis and diabetes) in both males and females.22,23
Accessible data for patients and health professionalsThe HI, in collaboration with national associations worldwide, has made available the data, with open access to patients and health professionals, including translations in several languages, of the cited “Therapeutic Recommendations” (for details, see at http://hemochromatosis-international.org/translations-therapeutic-recommendations).


