

Mutations in the globin genes that lead to amino acid substitutions in the globin chains result in hemoglobin (Hb) variants, and among these, hemoglobin S (Hb S) is the most well-known and frequent member worldwide, particularly in Brazil.1 Another variant is the Hb N-Baltimore (also known as Hb Jenkins, Hopkins-1, N-Memphis, and Kenwood), first described in 1958 by Smith and Torbert in an African-American family.2 This variant results from a mutation in codon 95 of the β globin chain, replacing the lysine amino acid for glutamic acid, which promotes a faster electrophoretic mobility of Hb N-Baltimore, in contrast with Hb A in alkaline pH.3 Using electrophoresis of hemoglobin at alkaline pH, high-performance liquid chromatography (HPLC) and sequencing procedures, we noticed an extremely rare interaction between the variant Hb S and Hb N-Baltimore in a patient admitted to a reference center in northeastern Brazil.
MethodsBlood samples were collected, with EDTA as the anticoagulant, from the child and family members after obtaining their informed consent. Hematological parameters were measured using a Coulter STKS (Coulter Electronics, Hialeah, FL, USA) and the erythrocyte morphology was analyzed. Biochemical parameters quantifications were performed by the Cobas C501 analyzer (Roche Diagnostics (®), Meylan, France). The methodological procedures applied included screening tests and validation for the diagnosis of hemoglobinopathy. Electrophoresis of hemoglobin at alkaline pH in cellulose acetate was executed, following which the Hb profile was investigated by cation-exchange high-performance liquid chromatography (Variant II™, Bio-Rad Laboratories, Hercules, CA, USA), as per the manufacturer's instructions. To confirm the presence of Hb S, the solubility test was executed.4 Sequencing procedures were performed on the ABI PRISM® 3500 Genetic Analyzer for identification of the unknown Hb.
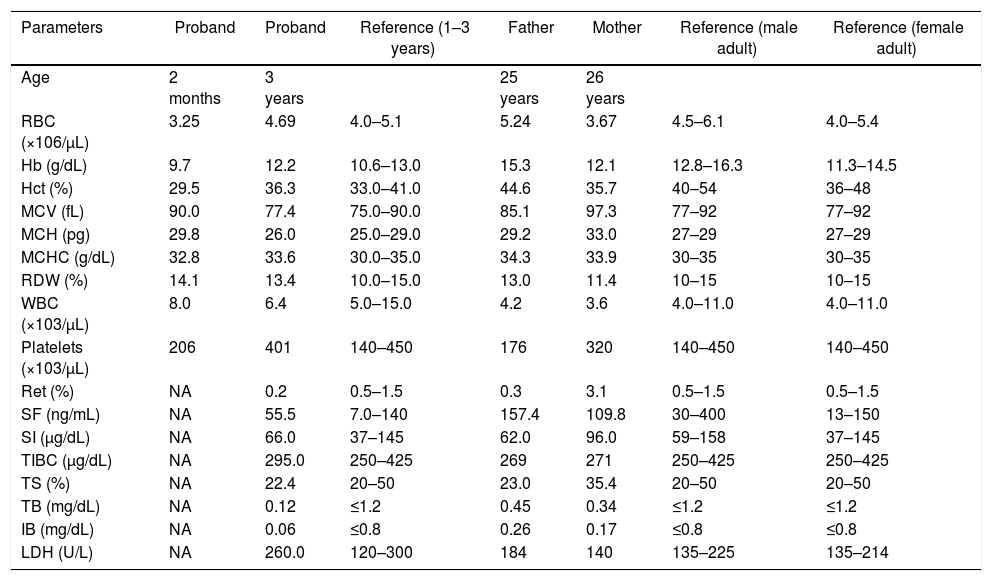
Case reportA 2-month-old female child was admitted to the Hospital of Hematology and Hemotherapy Foundation of Pernambuco in July 2015, after variant hemoglobin was identified during the newborn screening test. The peripheral blood analysis showed normocytic/normochromic anemia (Table 1). The electrophoresis of hemoglobin at alkaline pH revealed migration of a slower band than hemoglobin A (Hb A), which is suggestive of Hb S-like and was confirmed by a solubility test, and a faster band than Hb A, co-eluent to Hb J (Figure 1A). The quantification by cationic exchange HPLC, showed 56% of fetal hemoglobin (Hb F), 13% of Hb S, 2% of Hb A2, and 29% of unknown Hb (so-called Hb X), with a retention time of 1.58min (Figure 1B). Screening for hemoglobin variants through sequencing of the β globin gene revealed in heterozygosis a β95 Lys>Glu; HBB: c.286A>G, characterizing Hb N-Baltimore, along with the mutation β6 Glu>Val HBB: c.20A>T, corresponding to Hb S (Figure 1D). Further molecular analyses of the parents, both of African descent, showed the presence of Hb S in heterozygosis in the father, and Hb N-Baltimore, also in heterozygosis, in the mother.
Summary of hematologic and biochemical data of the patient and his family members.
| Parameters | Proband | Proband | Reference (1–3 years) | Father | Mother | Reference (male adult) | Reference (female adult) |
|---|---|---|---|---|---|---|---|
| Age | 2 months | 3 years | 25 years | 26 years | |||
| RBC (×106/μL) | 3.25 | 4.69 | 4.0–5.1 | 5.24 | 3.67 | 4.5–6.1 | 4.0–5.4 |
| Hb (g/dL) | 9.7 | 12.2 | 10.6–13.0 | 15.3 | 12.1 | 12.8–16.3 | 11.3–14.5 |
| Hct (%) | 29.5 | 36.3 | 33.0–41.0 | 44.6 | 35.7 | 40–54 | 36–48 |
| MCV (fL) | 90.0 | 77.4 | 75.0–90.0 | 85.1 | 97.3 | 77–92 | 77–92 |
| MCH (pg) | 29.8 | 26.0 | 25.0–29.0 | 29.2 | 33.0 | 27–29 | 27–29 |
| MCHC (g/dL) | 32.8 | 33.6 | 30.0–35.0 | 34.3 | 33.9 | 30–35 | 30–35 |
| RDW (%) | 14.1 | 13.4 | 10.0–15.0 | 13.0 | 11.4 | 10–15 | 10–15 |
| WBC (×103/μL) | 8.0 | 6.4 | 5.0–15.0 | 4.2 | 3.6 | 4.0–11.0 | 4.0–11.0 |
| Platelets (×103/μL) | 206 | 401 | 140–450 | 176 | 320 | 140–450 | 140–450 |
| Ret (%) | NA | 0.2 | 0.5–1.5 | 0.3 | 3.1 | 0.5–1.5 | 0.5–1.5 |
| SF (ng/mL) | NA | 55.5 | 7.0–140 | 157.4 | 109.8 | 30–400 | 13–150 |
| SI (μg/dL) | NA | 66.0 | 37–145 | 62.0 | 96.0 | 59–158 | 37–145 |
| TIBC (μg/dL) | NA | 295.0 | 250–425 | 269 | 271 | 250–425 | 250–425 |
| TS (%) | NA | 22.4 | 20–50 | 23.0 | 35.4 | 20–50 | 20–50 |
| TB (mg/dL) | NA | 0.12 | ≤1.2 | 0.45 | 0.34 | ≤1.2 | ≤1.2 |
| IB (mg/dL) | NA | 0.06 | ≤0.8 | 0.26 | 0.17 | ≤0.8 | ≤0.8 |
| LDH (U/L) | NA | 260.0 | 120–300 | 184 | 140 | 135–225 | 135–214 |
RBC: red blood cells; Hb: hemoglobin; Hct: hematocrit; MCV: mean corpuscular volume; MCH: mean corpuscular hemoglobin; MCHC: mean corpuscular hemoglobin concentration; RDW: red cell distribution width; WBC: white blood cells; Ret: reticulocyte count; SF: serum ferritin; SI: serum iron; TIBC: total iron binding capacity; TS: transferrin saturation; TB: total bilirubin; IB: indirect bilirubin; LDH: lactate dehydrogenase; NA: not available.
 Electrophoresis of hemoglobins at pH 8.0. 1: Hb SS, 2: Hb AA, 3: Hb SF, 4: Hb AJ (mother), 5: Hb AS (father), 6: Hb SJF↑ (patient at two months), 7: Hb AS. (B) HPLC of the patient at two months showing Hb F, unknown Hb, Hb A2 and Hb S. (C) HPLC of the patient at three years showing Hb F, unknown Hb, Hb A2 and Hb S. (D) Electropherogram of the β globin gene. Sequencing of the β globin gene revealed a mutation in the codon 95 (β95 Lys>Glu; HBB: c.286A>G) in heterozygosis, characterizing the Hb N-Baltimore variant, as well as a mutation in codon 6 (β6 Glu>Val HBB: c.20A>T) corresponding to Hb S.")
(A) Electrophoresis of hemoglobins at pH 8.0. 1: Hb SS, 2: Hb AA, 3: Hb SF, 4: Hb AJ (mother), 5: Hb AS (father), 6: Hb SJF↑ (patient at two months), 7: Hb AS. (B) HPLC of the patient at two months showing Hb F, unknown Hb, Hb A2 and Hb S. (C) HPLC of the patient at three years showing Hb F, unknown Hb, Hb A2 and Hb S. (D) Electropherogram of the β globin gene. Sequencing of the β globin gene revealed a mutation in the codon 95 (β95 Lys>Glu; HBB: c.286A>G) in heterozygosis, characterizing the Hb N-Baltimore variant, as well as a mutation in codon 6 (β6 Glu>Val HBB: c.20A>T) corresponding to Hb S.
Three years later, subsequent HPLC showed 7% of Hb F, 32.9% of Hb S, 2.8% of Hb A2 and 57.3% of Hb N-Baltimore, with a retention time of 1.58min (Figure 1C). Hematoscopy revealed erythrocyte morphology within the parameters of normality and the biochemical data was normal (Table 1). The child was asymptomatic, weighing 14kg, and measuring 100cm, with eutrophic nutritional status (BMI percentile=50), blood pressure at 105/71 and spleen not palpable, as well as dullness to percussion over the left intercostal Traube's space. To date, the child is annually followed up by the sickle cell disease (SCD) clinical program, but no SCD prophylactic therapy was indicated, as the patient is asymptomatic.
DiscussionThe Hb N-Baltimore results from a mutation in codon 95 of the β globin chain, replacing the lysine amino acid for glutamic acid β95 Lys>Glu; HBB: c.286A>G, which promotes a faster electrophoretic mobility, compared to the Hb A in alkaline pH, but does not present a difference, as to migration at acidic pH. In addition, Hb N-Baltimore has normal oxygen affinity and stability and an excellent separation by IEF (isoelectric focusing).3,5
A previous study showed that the interaction of Hb N-Baltimore with Hb S in three children in France resulted in no alteration of clinical symptoms and normal hematologic and biochemical parameters.6 This variant was also described in association with β-thalassemia and Hb C, and although Hb N-Baltimore accelerates Hb C crystallization and contributes to abnormalities in erythrocyte morphology, this combination leads to a mild clinical course in the carriers.7–9 Although it has no clinical relevance, little is known about the interaction between Hb N-Baltimore and Hb S.6 Thus, this report is the first to elucidate this interaction in South America. In the case we studied, the child is clinically healthy 3 years after diagnosis. Nevertheless, owing to the short clinical follow-up of the patient, long-term complications cannot be predicted. The clinically healthy status of the patient may lie in the protective effect of Hb F, as it prevents the Hb S polymerization; however, later analyses showed a decreasing percentage of Hb F and higher levels of the Hb N-Baltimore/Hb S ratio, which may presuppose a dominant effect of Hb N-Baltimore over the Hb S. This finding can be explained by the high stability of the Hb N-Baltimore,10 which presents similar biochemical features to Hb A, including the affinity for alpha globin chains.11 For this reason, possible intermolecular cooperation between the Hb S and Hb N-Baltimore variants should be further explored.
In summary, we herein describe a rare case of interaction between Hb N-Baltimore and Hb S in a health carrier, which was identified through electrophoretic analysis, HPLC and sequencing methodology. The association of these methodological procedures is useful in the identification of rare variant hemoglobins and aids in the avoidance of erroneous diagnoses, thus leading to an appropriate clinical approach. In addition, the study of these interactions enhances the knowledge on the pathophysiology of sickle cell disease.
Author contributionsAll authors read and approved the manuscript.
Conflicts of interestThe authors declare no conflicts of interest.



