


Haploinsufficiency of the hematopoietic transcription factor GATA2 is associated with a broad spectrum of diseases, including infection susceptibility and neoplasms. We aimed to investigate GATA2 variants in patients with non-tuberculous mycobacterial (NTM) and/or fungal infections (FI) without known immunodeficiencies.
MethodWe performed GATA2 genotyping in patients with NTM and/or FI.
ResultsTwenty-two patients were enrolled (seventeen FI, four NTM and one with both infections). The pathogenic variant NG_029334.1:g.16287C>T was found in one patient (4.5%) and two asymptomatic offsprings. We also found the likely-benign variant NG_029334.1:g.12080G>A (rs2335052), the benign variant NG_029334.1:g.16225C>T (rs11708606) and the variant of uncertain significance NG_029334.1:g.16201G>A (rs369850507) in 18.2%, 27.3%, and 4.5% of the cases, respectively. Malignant diseases were additionally diagnosed in six patients.
ConclusionAlthough detected in 45.4% of the patients, most GATA2 variants were benign or likely benign. Identifying a pathogenic variant was essential for driving both the patient's treatment and familial counseling. Pathogenic variants carriers should receive genetic counseling, subsequent infection prevention measures and malignancies surveillance. Additionally, case-control genotyping should be carried out in Brazil to investigate whether the observed variants may be associated with susceptibility to opportunistic infections and/or concurrent neoplasms.
Non-tuberculous mycobacteria (NTM) are ubiquitous in soil, water and human-made environments. Infections caused by these organisms seem to be increasing worldwide and the immune host defects that may underly it were revised in 2015.1 Advances in imaging, microbiological and molecular techniques have contributed to recognizing these infections, leading to an apparent increased incidence rate over time.2 Morbidity caused by NTM is clinically manifested by lymphadenitis, skin and pulmonary diseases. The outcome of NTM infection depends upon a complex interaction between exposure, duration of host exposure, immune status and genetic background.3 Overall, whether primary or acquired, patients with immunodeficiencies are at risk of developing NTM or fungal infections (FI).4 Currently, recognizing the underlying genetic background for NTM and/or FI is essential for therapy and prognostic assessment. Some genetic variants in interleukin 12-interferon γ pathway genes (INGR1/2, IL12b, STAT1) are often associated with the onset of opportunistic infections in early childhood. In contrast, GATA2 pathogenic genetic variants give rise to disseminated infection in late childhood or adulthood.1
The GATA2 gene is located on the chromosome 3q21 region, encoding a 50.5 KDa protein. The GATA2 has six exons and two zinc-finger domains and it acts as a transcription factor in several tissues.5 The GATA2 is mostly expressed in hematopoietic progenitors, myeloid and mast cells.6 The GATA2 haploinsufficiency has been associated with the MonoMAC syndrome, characterized by a decreased number of monocytes, T, B and NK cells, infections, autoimmunity and pulmonary alveolar proteinosis in both sporadic and familial forms. Additionally, the GATA2 haploinsufficiency has been associated with lymphedema, warts and deafness, a condition named Emberger syndrome. Both MonoMAC and Emberger syndromes predispose patients to myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML).5 Because of the importance of identifying the underlying causes of overt immunodeficiency in patients with NTM and/or FI, this study aimed to identify GATA2 genetic variants among such individuals and establish associations with clinical-phenotypic features.
MethodsA series of patients older than 12 years old with pulmonary or disseminated NTM and/or FI were enrolled between 2015 to 2018 at the Instituto Nacional de Infectologia Evandro Chagas, Fundação Oswaldo Cruz (FIOCRUZ), Rio de Janeiro, Brazil. We used convenience sampling for patients recruited during hematological consultations. Demographic, clinical and laboratory features were obtained from the patients' medical records and during follow-up. A complete blood count was performed upon patient inclusion in the study (Supplementary Table 1). The human immunodeficiency virus (HIV) serology was tested in all patients. The infectious diagnosis was confirmed with direct microscopic examination, staining for acid-fast bacillus (AFB), histopathological and/or microbiological culture analysis, when available. Bone marrow assessment was performed for a myelogram, immunophenotyping, histopathological and cytogenetics examination in patients who presented sustained unexplained cytopenias or organomegalies (Supplementary Table 2).
The exclusion criteria were the prior diagnosis of primary or secondary immunodeficiencies, such as acquired immunodeficiency syndrome/HIV infection; well established autoimmune diseases, such as rheumatoid arthritis; solid organ transplantation; cystic fibrosis, and; immunosuppressive or chemotherapeutic treatments.
According to the manufacturer's instructions, genomic DNA was extracted from peripheral blood samples using the commercial QIAamp DNA Blood Mini Kit (Qiagen, CA, USA). Sanger sequencing was performed throughout the five coding exons (2, 3, 4, 5 and 6) and intron 4 of the GATA2 gene using the protocol described by Wang et al.7 Whenever GATA2 pathogenic variants were identified in the proband, they were also investigated in the relatives. All PCR products were sequenced using the ABI3500xl automatic gene analyzer (Applied Biosystems, CA, USA). Sequences were analyzed in the Mutation Surveyer® software (Softgenetics, PA, USA). The GATA2 genetic variants identified were compared to the reference databases available online at https://cancer.sanger.ac.uk/cosmic, https://www.ncbi.nlm.nih.gov/clinvar and www.ensemble.org to evaluate the clinical association. The nomenclature was applied following the American College of Medical Genetics and Genomics and the Association for Molecular Pathology guidelines.8 We classified variants into the five categories "pathogenic," "likely pathogenic," "variant of uncertain significance (VUS)," "likely benign," and "benign". The terms "likely pathogenic" and "likely benign" are used to mean greater than 90% certainty of a variant either being disease-causing or benign, respectively.8 Frequency analyses were performed with the IBM® SPSS Statistics version 22.0.
EthicsPatients were treated following the Declaration of Helsinki and the Brazilian ethical standards – Resolution 466/12. Written informed consent was obtained from the individuals to participate in this research. The Review Board of the Evandro Chagas National Institute of Infectious Diseases approved the study (#64574417.0.0000.5262).
ResultsTwenty-seven patients fulfilled the study inclusion criteria, but five patients declined the invitation to participate. Thirty-one samples from 22 patients and nine healthy relatives from a single family were genotyped. There was a predominance of males and white patients (63.6% and 59.1%, respectively). The median age was 45.5 years old (range 19 - 75 years). The most frequent infection groups were FI (77.3%), followed by NTM (18.2%) and one patient had both NTM and FI (4.5%). The study design and main findings are illustrated in Figure 1.
 and fungal infection (FI) cases were identified, five were excluded, as they declined to participate, leaving 22 eligible cases. From these, four were NTM, 17 were FI and one case presented both infections. The GATA2 genotyping was performed in all cases and four variants were identified. The NG_029334.1:g.16287C>T, a pathogenic variant, was detected in 1 NTM case and two relatives (out of nine relatives tested). The likely-benign variant NG_029334.1:g.12080G>A (rs2335052) was identified in 2 NTM cases, one of which also carried the benign variant NG_029334.1:g.16225C>T (rs11708606) and in 2 FI cases. The benign variant NG_029334.1:g.16225C>T (rs11708606) was detected in the simultaneous NTM and FI case and in 4 FI cases, one of which also carried the variant of uncertain significance NG_029334.1:g.16201G>A (rs369850507).")
Schematic representation of the study design and main findings. From 2015 to 2018, 27 non-tuberculous mycobacteria (NTM) and fungal infection (FI) cases were identified, five were excluded, as they declined to participate, leaving 22 eligible cases. From these, four were NTM, 17 were FI and one case presented both infections. The GATA2 genotyping was performed in all cases and four variants were identified. The NG_029334.1:g.16287C>T, a pathogenic variant, was detected in 1 NTM case and two relatives (out of nine relatives tested). The likely-benign variant NG_029334.1:g.12080G>A (rs2335052) was identified in 2 NTM cases, one of which also carried the benign variant NG_029334.1:g.16225C>T (rs11708606) and in 2 FI cases. The benign variant NG_029334.1:g.16225C>T (rs11708606) was detected in the simultaneous NTM and FI case and in 4 FI cases, one of which also carried the variant of uncertain significance NG_029334.1:g.16201G>A (rs369850507).
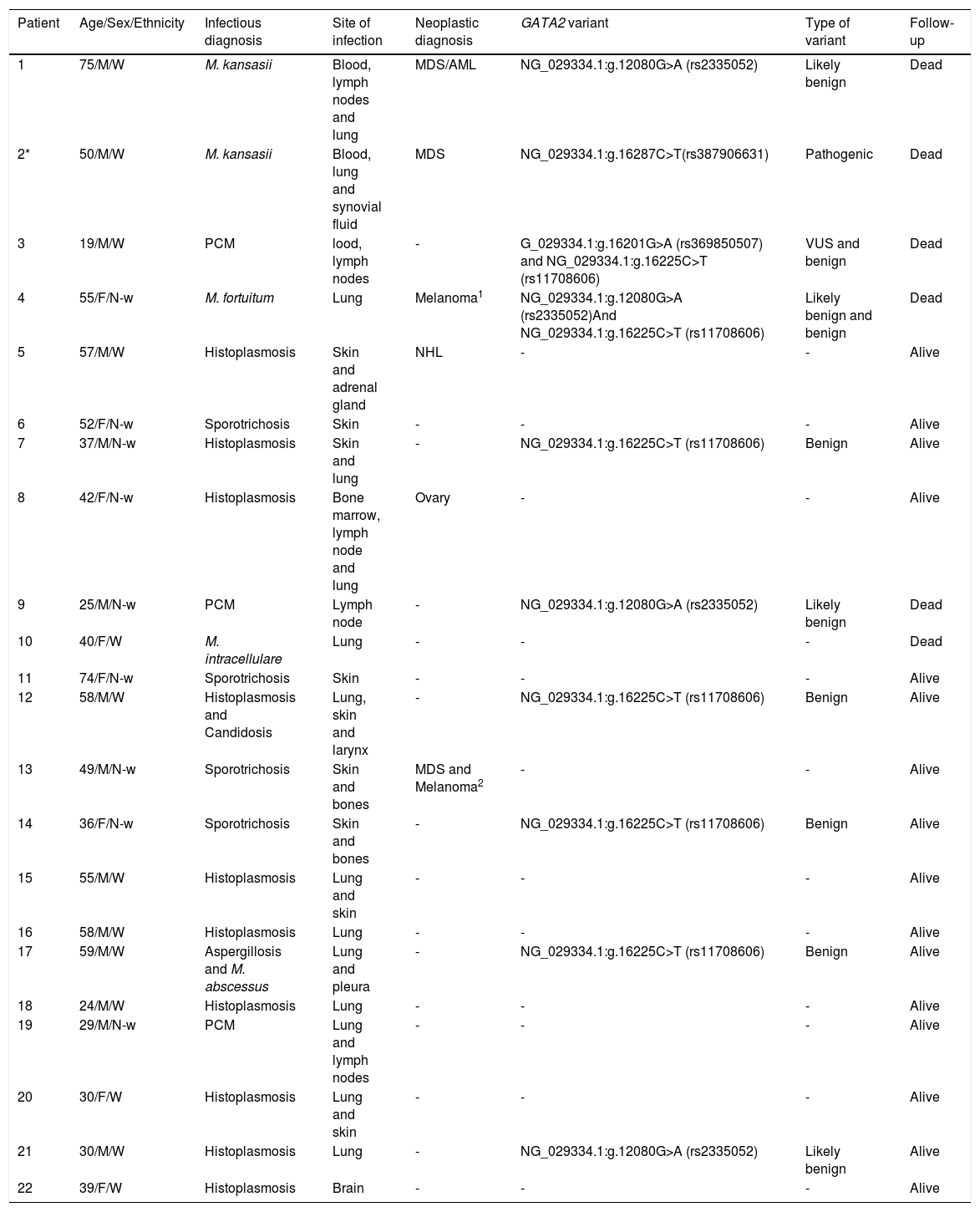
Among the FIs, histoplasmosis, sporotrichosis, paracoccidioidomycosis, aspergillosis and candidosis were detected. The NTM cases were caused by Mycobacterium kansasii, M. abscessus, M. fortuitum and M. intracellulare. The infections were identified at the following sites: blood, lymph node, lung, synovial fluid, skin, adrenal gland, larynx, bone marrow, bones, pleura and brain. The demography, clinical information and GATA2 variants are depicted in Table 1.
Summary of main clinical findings and GATA2 genetic variants in the ascertain patients.
| Patient | Age/Sex/Ethnicity | Infectious diagnosis | Site of infection | Neoplastic diagnosis | GATA2 variant | Type of variant | Follow-up |
|---|---|---|---|---|---|---|---|
| 1 | 75/M/W | M. kansasii | Blood, lymph nodes and lung | MDS/AML | NG_029334.1:g.12080G>A (rs2335052) | Likely benign | Dead |
| 2* | 50/M/W | M. kansasii | Blood, lung and synovial fluid | MDS | NG_029334.1:g.16287C>T(rs387906631) | Pathogenic | Dead |
| 3 | 19/M/W | PCM | lood, lymph nodes | - | G_029334.1:g.16201G>A (rs369850507) and NG_029334.1:g.16225C>T (rs11708606) | VUS and benign | Dead |
| 4 | 55/F/N-w | M. fortuitum | Lung | Melanoma1 | NG_029334.1:g.12080G>A (rs2335052)And NG_029334.1:g.16225C>T (rs11708606) | Likely benign and benign | Dead |
| 5 | 57/M/W | Histoplasmosis | Skin and adrenal gland | NHL | - | - | Alive |
| 6 | 52/F/N-w | Sporotrichosis | Skin | - | - | - | Alive |
| 7 | 37/M/N-w | Histoplasmosis | Skin and lung | - | NG_029334.1:g.16225C>T (rs11708606) | Benign | Alive |
| 8 | 42/F/N-w | Histoplasmosis | Bone marrow, lymph node and lung | Ovary | - | - | Alive |
| 9 | 25/M/N-w | PCM | Lymph node | - | NG_029334.1:g.12080G>A (rs2335052) | Likely benign | Dead |
| 10 | 40/F/W | M. intracellulare | Lung | - | - | - | Dead |
| 11 | 74/F/N-w | Sporotrichosis | Skin | - | - | - | Alive |
| 12 | 58/M/W | Histoplasmosis and Candidosis | Lung, skin and larynx | - | NG_029334.1:g.16225C>T (rs11708606) | Benign | Alive |
| 13 | 49/M/N-w | Sporotrichosis | Skin and bones | MDS and Melanoma2 | - | - | Alive |
| 14 | 36/F/N-w | Sporotrichosis | Skin and bones | - | NG_029334.1:g.16225C>T (rs11708606) | Benign | Alive |
| 15 | 55/M/W | Histoplasmosis | Lung and skin | - | - | - | Alive |
| 16 | 58/M/W | Histoplasmosis | Lung | - | - | - | Alive |
| 17 | 59/M/W | Aspergillosis and M. abscessus | Lung and pleura | - | NG_029334.1:g.16225C>T (rs11708606) | Benign | Alive |
| 18 | 24/M/W | Histoplasmosis | Lung | - | - | - | Alive |
| 19 | 29/M/N-w | PCM | Lung and lymph nodes | - | - | - | Alive |
| 20 | 30/F/W | Histoplasmosis | Lung and skin | - | - | - | Alive |
| 21 | 30/M/W | Histoplasmosis | Lung | - | NG_029334.1:g.12080G>A (rs2335052) | Likely benign | Alive |
| 22 | 39/F/W | Histoplasmosis | Brain | - | - | - | Alive |
M: Male; F: Female; W: White; N-w: Non-white; FH: Familial history; PCM: Paracoccidioidomycosis; NTM: Nontuberculous mycobacteria; MDS: Myelodysplastic syndrome; AML: Acute myeloid leukemia; NHL: Non-Hodgkin Lymphoma of skin; VUS: Variant of uncertain significance.
MAF: Minor allele frequency in the population;
Single nucleotide variants were fund in 10 out of 22 patients (45.4%), with exons 3 and 5 harboring the most frequent genetic alterations. Four genetic variants were identified. Only one patient (4.5%) exhibited the pathogenic loss-of-function missense variant NG_029334.1:g.16287C>T. It was identified in a 43 years-old white male with a 20-year follow-up of autoimmune and thrombotic phenomena, hypothyroidism, disseminated refractory M. kansasii infection, MonoMAC syndrome and a progressive MDS. The same mutation was detected in two asymptomatic offsprings out of 9 relatives tested. His complete clinical presentation and pedigree were reported elsewhere.9 Four patients (18.2%) presented the likely benign variant NG_029334.1:g.12080G>A (rs2335052) in exon 3. We also identified the benign variant NG_029334.1:g.16225C>T (rs11708606) in 6 patients (27.3%), and the VUS NG_029334.1:g.16201G>A (rs369850507) in one patient (4.5%). According to the ALFA Project consulted at https://www.ncbi.nlm.nih.gov on 09/01/20, the minor allele frequency of the variants NG_029334.1:g.12080G>A (rs2335052), NG_029334.1:g.16225C>T (rs11708606) and NG_029334.1:g.16201G>A (rs369850507) were 16.5%, 16.5% and 0.01% respectively, among the total general population.
Six patients developed neoplasms either before or after the infectious disease diagnosis, as described in Table 1. Nevertheless, they had not received any immunosuppressive treatment before the study. Three presented with MDS, two with melanoma (one patient presented MDS and melanoma simultaneously), one with diffuse B-cell non-Hodgkin lymphoma and one patient with ovarian cancer. Two cases (one melanoma and the ovarian cancer patient) underwent prior surgery and the others received chemotherapy after enrollment. A mortality rate of 27.3% (6 patients) was observed. Three in 17 patients had low CD4+ lymphocyte levels and overall, three individuals presented monocytopenia (13.6%) and two of them suffered from MDS (see Supplementary Table 1).
DiscussionSince the first description of GATA2 haploinsufficiency was associated with primary immunodeficiency in 2011, 343 coding variants have been described by ClinGen/ClinVar and 168 are deemed pathogenic.10 So far, there are more than 100 families and 122 individual cases (7 pediatric cases of confirmed de novo) of GATA2 haploinsufficiency described.11 A new class of GATA2 germline mutation with the insertion of amino acids between the first and second zinc fingers was recently reported in Brazil.12 Herein, we present the prevalence of GATA2 genetic variants in Brazilian patients with NTM and/or FI without known immunodeficiencies. Overall, nine patients (40.9%) carried at least one of the GATA2 variants benign, likely benign or of uncertain significance and one individual presented a pathogenic variant with an NTM infection.
The NG_029334.1:g.16287C>T is one of the GATA2 pathogenic variants most frequently associated with the MonoMAC syndrome and is located at the second zinc finger domain.13 We identified this variant, also known as T354M, in a patient presenting NMT, MDS, monocytopenia and low CD4+ levels. Identifying it was essential for driving both the patient's treatment and familial counseling.9 The NG_029334.1:g.12080G>A (rs2335052) is not a pathogenic variant and does not affect the zinc finger domains. However, it is located in a coding and regulatory region. It has been described as an expression quantitative trait locus by the Genotype-Tissue Expression (GTEx) Project, contributing to the GATA2 expression regulation. Furthermore, this variant has been previously associated with a worse prognosis in a colon cancer study14 and described in an AML case report, along with a novel GATA2 pathogenic variant.15 The other variants identified here, the NG_029334.1:g.16225C>T (rs11708606) and NG_029334.1:g.16201G>A (rs369850507), encompass intronic regions and have not been explored in vitro. Although these non-pathogenic variants are not likely to exert a high penetrance effect on the disease onset, they might be associated with infection and/or progression susceptibility. Future case-control studies should explore this hypothesis. So far, the benign and VUS variants identified in this study are not clinically actionable and should not drive clinical management change.
The marked heterogeneity of the GATA2 haploinsufficiency clinical presentation is a diagnostic challenge. Infections, such as NTM or FI due to immune dysfunction, can be the first manifestation, persist throughout the patient's life and be a significant cause of death.16 A study observed disseminated NTM infections in 53% of the patients, including both slow growing (Mycobacterium avium complex, M. kansasii and M. szulgai) and rapid-growing mycobacteria (M. fortuitum, M. abscessus and M. chelonae), while severe FI were observed in 16%: invasive aspergillosis in 9%, disseminated histoplasmosis in 5% and recalcitrant mucosal candidiasis in 5%.17 However, some carriers can be asymptomatic or present just slight laboratory abnormalities. Despite the autosomal dominant inheritance, pathogenic variants can deliver a highly variable penetrance and many functional paths affecting the GATA2 expression are still unknown.5,10,16 The penetrance of the GATA2 haploinsufficiency increases with age, but remains incomplete at the age of 40 years old in 32.9% of the carriers.18 Moreover, few cases over 60 years old are asymptomatic.16,18 Epigenetic plasticity and coexisting somatic mutations can trigger pathogenesis and dictate penetrance.10 Catto et al. demonstrated somatic genetic rescue in the germline GATA2 haploinsufficiency setting.19 This finding would explain the early recovery of hematopoiesis and lack of symptoms in a mosaic 61-year-old carrier and a possible false-negative initial result.19 The MDS is the most common first malignancy seen in GATA2 carriers, with a noticeable peak in the second decade of life.11 The allogeneic stem cell transplant is available for the GATA2 haploinsufficiency in selected cases and the eligible related donors should be systematically tested.17,20
This study reinforces the need for multidisciplinary care for patients with oportunistic infections without known immunodeficiencies, particularly NMT with MDS, considering a putative germline GATA2 haploinsufficiency. Despite being associated with the susceptibility to FI, as well as being the majority of cases, we did not find any GATA2 pathogenic variant in our fungal cohort. Finally, we had limitations in this small, single-center and descriptive work; in particular, we did not perform a more comprehensive study of the immune competence and we could not rule out other causes of primary immunodeficiencies because we did not investigate such conditions.
ConclusionThe GATA2 genetic variants are found in patients presenting with NTM and/or FI without known immunodeficiency. The NTM/FI can indicate a primary immunodeficiency and some immunodeficiency patients are at risk of cancers, such as MDS and AML. Individuals with NMT infections and monocytopenia are suspected of having the MDS and MonoMac syndrome. Even asymptomatic carriers of pathogenic variants should receive genetic counseling, subsequent infection prevention measures and surveillance for hematologic malignancies. Further studies on the prevalence, expression and clinical significance of GATA2 variants are necessary to comprehend this condition in all its complexity and to be able to indicate the best and safest treatment in each case.
FundingThe MSPO and RMZO are supported by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) [#310877/2019-9 and #302796/2017-7] and by the Fundação de Amparo à Pesquisa do estado do Rio de Janeiro (FAPERJ) [E-26/203.076/2016 to RMZO]. SCSL is a scholar of FAPERJ [#E-26/202.918/2019].
Author's contributionsDPMA designed the study, attended to the patients, collected and analyzed the clinical data. DPMA, SCSL and MSPO wrote the manuscript; DPMA, FGA and FVSB performed the genetic tests; DFSF attended to patients and revised the article; MSPO and RMZO designed, provided funding and supervised the study. All authors have seen and approved the manuscript and its submission.
The authors thank the patients and relatives for making samples available and the physicians for providing clinical details, particularly Dr. Valeria Rolla. We also thank Alexandre Vizzoni for the laboratory and data set support and Prof. Logan Spector for the critical revision of the manucript. The authors are also grateful to the INI-FIOCRUZ Flow Cytometry platform, the Oswaldo Cruz Institute (IOC-FIOCRUZ) and the INCA sequencing platform for technical support on genetic testing. Finally, we thank the Genotype-Tissue Expression (GTEx) Project, supported by the Common Fund of the Office of the Director of the National Institutes of Health and by NCI, NHGRI, NHLBI, NIDA, NIMH and NINDS. The data used for the analyses described in this manuscript were obtained from the GTEx Portal on 08/23/20.


