

Human aplastic anemia is a hematologic disease characterized by low peripheral blood cell counts associated with reduced numbers of hematopoietic stem and progenitor cells and a hypocellular bone marrow. Thrombopoietin (THPO) regulates megakaryocytes, but it also stimulates hematopoietic stem and progenitor cells. Biallelic mutations in the THPO gene have been reported in a family with recessive inherited aplastic anemia.
MethodsThis study screened 83 patients diagnosed with acquired aplastic anemia and 92 paired healthy controls for germline variants in the THPO gene using Sanger sequencing.
ResultsThree common single nucleotide polymorphisms were identified in patients and controls at comparable allele frequencies. There was no correlation between the single nucleotide polymorphism carrier status and platelet counts at diagnosis.
ConclusionThe presence of THPO polymorphisms is comparable between patients with acquired aplastic anemia and healthy individuals.
Aplastic anemia (AA) is a hematologic disorder caused by hematopoietic stem cell failure, which clinically results in a hypocellular bone marrow and reduced blood cell counts.1,2 In humans, AA is rare, affecting approximately 1–2 patients per million inhabitants3 with the highest incidences in East Asian countries.1 In acquired AA, hematopoiesis is suppressed by an immune-mediated mechanism in which cytotoxic T-cells target hematopoietic stem and progenitor cells in the marrow.1 Other cases are inherited, and loss-of-function mutations in genes regulating DNA repair are etiologic.4
Thrombopoietin (THPO), a hormone produced in the liver, is a primary regulator of megakaryocyte expansion and differentiation and is vital for hematopoietic stem cell maintenance, working as a pan-hematopoietic cytokine.5 Loss-of-function mutations in THPO and in MPL, which encodes the thrombopoietin receptor MPL, are associated with a spectrum of marrow failures, from amegakaryocytic thrombocytopenia to familial AA.6 Biallelic mutations in the THPO gene were identified in a family with inherited AA.7 Conversely, gain-of-function mutations in these genes are observed in myelofibrosis, further implicating the THPO-MPL pathway in the regulation of hematopoietic stem cells.8
This study investigated whether germline mutations in the THPO gene were associated with acquired AA.
MethodsPatients and controlsDNA was extracted from bone marrow or peripheral blood cells as previously described.9 Samples were collected after informed consent, according to the protocol approved by the local Research Ethics Committee (CEP-HCRP, protocol number 518/2015). Patients diagnosed with AA at the hematology clinic of the Universidade Federal do Paraná (UFPR) for whom stored DNA was available were eligible for the study. No participant underwent hematopoietic stem cell transplantation prior to sample collection. AA was diagnosed based on standard criteria.10,11 Patients with Fanconi anemia, dyskeratosis congenita, or Shwachman-Diamond syndrome were excluded. Age and gender-matched healthy blood donors were studied as healthy controls.
Sanger sequencingPrimers were designed by the Primer 3 program (web version 4.0.0), using the sequence Homo sapiens thrombopoietin (THPO) transcription variant 1, mRNA of NCBI with reference number NM_000460.3. Exon 6 was divided into two amplification reactions. Primer sequences are detailed in Table S1. The final reagent concentration in each polymerase chain reaction (PCR) was 100ng DNA, 0.2mM of each dNTP, 1.5mM of MgCl2, 1U of Taq DNA Polymerase, buffer 1×, 150nM of each primer (forward and reverse), in a final reaction volume of 25μL. The PCR conditions used for the assays were 5min at 95°C for initial denaturing, and 35 cycles at 95°C for 40s, 55 to 62°C (depending on the primer) for 45s for annealing and 72°C for 45s for extension.
After the PCR, amplicons were purified and sequenced using the BigDye Terminator Cycle Sequencing Kit (Applied Biosystems), with initial denaturing at 96°C for 2min, and 35 cycles at 95°C for 10s, 51°C for 10s and 60°C for 4min. The sequences were processed by the ABI 3500 XL detection system (Applied Biosystems) and analyzed with the Sequencher software (Gene Codes Corporation).
Statistical analysisPearson's chi-square test was used to compare the frequency of variants in patients with acquired AA (study group) and healthy individuals (control). A 5% level of statistical significance was considered for all the analyses. The IBM Statistical Package for Social Sciences (SPSS) version 19.0 was used.
ResultsClinical featuresFifty-nine percent of patients were male, and the mean age was 25±17 years. Patients’ demographics and blood counts at the time of diagnosis are provided in Table 1.
Demographic and clinical features of patients with acquired aplastic anemia (n=83).
| Characteristic | Value |
|---|---|
| Age – years | |
| Mean | 25 |
| Range | 2–74 |
| Gender – n | |
| Male | 49 |
| Race/ethnicity – n | |
| White | 57 |
| Mixed race | 13 |
| Black | 12 |
| Asian | 1 |
| Peripheral blood counts – mean±SD | |
| Hemoglobin (g/dL) | 7.2±0.4 |
| Hematocrit (%) | 20.8±3.1 |
| Neutrophils (/μL) | 188±158 |
| Platelets (/μL) | 2500±707 |
| Disease classification – n | |
| Moderate | 14 |
| Severe | 20 |
| Very severe | 49 |
| Treatment regimena– n | |
| Cyclosporine | 69 |
| Anti-thymocyte globulin | 20 |
| Androgen | 12 |
| Erythropoietin | 3 |
| Immunoglobulin | 3 |
| No treatment | 2 |
| Death – n | |
| Yes | 25 |
| No | 58 |
SD: standard deviation.
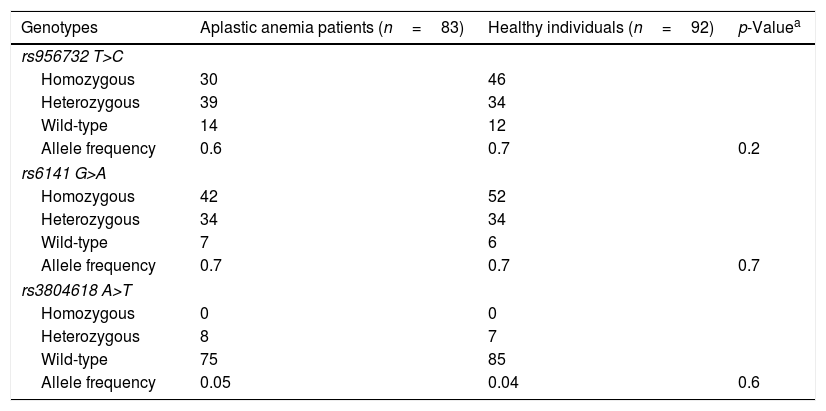
Three synonymous single nucleotide polymorphisms (SNPs) in the THPO gene were identified in patients and controls (Table 2). The allele frequencies for the three SNPs were comparable between patients and controls. No novel or rare variant was identified.
Single nucleotide polymorphisms in the patients with acquired aplastic anemia (study group) and healthy individuals (control group).
| Genotypes | Aplastic anemia patients (n=83) | Healthy individuals (n=92) | p-Valuea |
|---|---|---|---|
| rs956732 T>C | |||
| Homozygous | 30 | 46 | |
| Heterozygous | 39 | 34 | |
| Wild-type | 14 | 12 | |
| Allele frequency | 0.6 | 0.7 | 0.2 |
| rs6141 G>A | |||
| Homozygous | 42 | 52 | |
| Heterozygous | 34 | 34 | |
| Wild-type | 7 | 6 | |
| Allele frequency | 0.7 | 0.7 | 0.7 |
| rs3804618 A>T | |||
| Homozygous | 0 | 0 | |
| Heterozygous | 8 | 7 | |
| Wild-type | 75 | 85 | |
| Allele frequency | 0.05 | 0.04 | 0.6 |
The three SNPs are located in untranslated regions (UTR) of the THPO exons and therefore not represented in the THPO translation. The SNP rs956732 is located in the 5′ UTR of isoform 1 (NM_000460.3), and leads to a synonymous substitution in isoform 5 (NM_001290003.1). The SNPs rs6141 and rs3804618 are located in the 3′ UTR of the THPO mRNA. According to the ClinVar database (of the NCBI), the rs956732, rs6141, and rs3804618 SNPs have a benign clinical significance and are observed in allele frequencies of 0.5, 0.4, and 0.07, respectively, in the population, which is similar to our findings (Table 2).
As the THPO gene regulates thrombopoiesis, we investigated whether the THPO SNPs correlated with platelet levels in patients; no association was found between SNP carrier status and platelet counts (data not shown).
DiscussionThis study screened 83 patients with acquired AA and 92 matched controls for variants in the THPO gene. Three common SNPs were identified, but their prevalences were comparable between patients and controls. SNPs did not correlate with platelet counts in patients.
The THPO-MPL pathway regulates hematopoietic stem cell maintenance, and loss-of-function mutations in the THPO gene have been previously described in patients with familial AA. More recently, THPO mutations were found in five AA patients who underwent hematopoietic stem cell transplantation but failed to show engraftment.12 Two instances were treated with eltrombopag, an MPL agonist, resulting in effective engraftment. Together, these findings suggest that pathogenic mutations in the THPO gene may be etiologic in a minority of AA cases. Clinically, the addition of eltrombopag to standard immunosuppressive therapy as the first-line treatment of acquired AA resulted in a significantly better hematologic response in up to 90% of patients and patient survival in comparison to historic controls.13 When eltrombopag was added to horse antithymocyte globulin and cyclosporine, patients showed faster recovery of peripheral blood counts and normalization of the CD34+ cell compartment in the bone marrow. In patients with refractory acquired AA, monotherapy with eltrombopag achieved hematologic response in 44% of cases.14 In aggregate, these findings implicate the THPO-MPL pathway as a therapeutic target in acquired AA.
We asked the question whether genetic lesions would modulate THPO in acquired AA, since a previous study reported one mutation (THPO R17C) in familial AA.7 However, this study did not identify pathogenic variants in patients, and the allele frequencies of three common SNPs were similar between patients and controls.
In conclusion, pathogenic variants in the THPO gene are not associated with acquired AA.
Conflict of interestThe authors declare no conflicts of interest.
This study was supported by the São Paulo Research Foundation (FAPESP) grant 13/08135-2. FS Donaires received a FAPESP's post-doctoral fellowship (17/09428-4).
The following are the supplementary data to this article:



