




Daratumumab is a CD38-targeting monoclonal antibody with established efficacy and safety in patients with relapsed or refractory multiple myeloma (RRMM). We report results of an early access protocol (EAP) of daratumumab monotherapy for RRMM in a cohort of Brazilian patients.
MethodsPatients with RRMM and ≥3 prior lines of therapy, including a proteasome inhibitor (PI) and an immunomodulatory drug (IMiD), or who were double refractory to both a PI and IMiD received daratumumab, 16 mg/kg, intravenously weekly for 8 weeks, biweekly for 16 weeks, and every 4 weeks thereafter until disease progression, unacceptable toxicity, loss of clinical benefit, or study conclusion or if daratumumab became available with reimbursement.
ResultsForty-nine patients received ≥1 dose of daratumumab. The median (range) duration of treatment was 6.4 (0.3−11.8) months, with a median (range) of 8 (1−13) treatment cycles. Grade 3/4 treatment-emergent adverse events (TEAEs) were reported in 38.8% of patients, most frequently neutropenia and pneumonia (10.2% each). Seven (14.3%) patients discontinued treatment due to TEAEs; 3 patients discontinued due to daratumumab-related TEAEs. Serious TEAEs occurred in 38.8% of patients. Infusion-related reactions were reported in 25 (51.0%) patients, were primarily grade 1/2, and the majority (23 patients) occurred during the first infusion. Twenty (40.8%) patients achieved a partial response or better; median progression-free survival was 8.25 (95% confidence interval, 5.55–17.54) months.
ConclusionIn this EAP, daratumumab monotherapy in Brazilian patients showed a safety and efficacy profile consistent with clinical studies of daratumumab monotherapy in patients with heavily pretreated RRMM.
ClinicalTrials.gov identifier: NCT02477891.
Over the past 2 decades, 5-year survival rates for patients with multiple myeloma (MM) have markedly improved in the United States and Europe with the introduction of novel immunomodulatory drugs (IMiDs; eg, thalidomide and lenalidomide) and proteasome inhibitors (PIs; eg, bortezomib and carfilzomib).1–3 In Brazil and other Latin American countries, similar survival trends have been reported for patients with MM4; however, access to new treatments may be delayed due to cost concerns or resource limitations.5,6 The efficacy of MM treatment is substantially reduced following the first line of therapy, and progression-free survival (PFS) is poor among patients with relapsed or refractory MM (RRMM).5 Access to novel therapies and improvement of outcomes among Latin American patients with RRMM remains an unmet need.
Daratumumab is a human IgGκ monoclonal antibody targeting CD38 with a direct on-tumor7–10 and immunomodulatory11–13 mechanism of action. The efficacy and safety of daratumumab (16 mg/kg intravenous) monotherapy in patients with heavily pretreated RRMM was investigated in the open-label phase 1/2 GEN501 and phase 2 SIRIUS studies after median durations of follow-up of 10.2 months and 9.3 months, respectively.14,15 In a combined analysis of GEN501 and SIRIUS (median follow-up: 20.7 months), daratumumab achieved an overall response rate of 31.1%, median duration of response was 7.6 months, and median overall survival was 20.1 months.16 Daratumumab demonstrated a tolerable safety profile with no new safety concerns identified with longer follow-up.16 The addition of daratumumab to standard-of-care regimens in subsequent phase 3 clinical trials demonstrated superior clinical efficacy versus standard of care alone across lines of therapy, including patients with RRMM who have received at least 1 prior line of therapy,17,18 as well as patients with newly diagnosed MM (NDMM) who are eligible or ineligible for autologous stem cell transplant.19–21 Based on the results of these studies, daratumumab is approved for use in many countries as monotherapy for heavily pretreated RRMM and in combination with standard-of-care regimens for NDMM and RRMM.22 Daratumumab was approved in Brazil as monotherapy for heavily pretreated RRMM and in combination with bortezomib/dexamethasone for patients with RRMM with at least 1 prior line of therapy in January 2017. Subsequently, daratumumab was approved in Brazil in combination with lenalidomide/dexamethasone for patients with RRMM with at least 1 prior line of therapy, in combination with lenalidomide/dexamethasone or bortezomib/melphalan/prednisone for transplant-ineligible patients with NDMM, and in combination with bortezomib/thalidomide/dexamethasone for transplant-eligible patients with NDMM.6
Despite the established benefit of daratumumab in patients with RRMM and NDMM, not all patients are eligible for inclusion in ongoing daratumumab clinical trials or have access to commercially available daratumumab. MMY3010 is a multicenter, open-label early access treatment protocol (EAP; ClinicalTrials.gov Identifier: NCT02477891) with the objective of providing early access to daratumumab monotherapy for eligible patients with heavily pretreated RRMM while collecting additional safety data. Results from MMY3010 patient cohorts from the United States, Spain, Italy, Russia, and the United Kingdom were consistent with the GEN501 and SIRIUS clinical trials of daratumumab monotherapy and confirmed the safety profile of daratumumab in patients with heavily pretreated RRMM.14,15,23,24 Here, we present findings from the Brazilian cohort of MMY3010.
MethodsPatientsPatients were aged ≥18 years with documented MM and evidence of disease progression on or after the most recent prior treatment regimen as defined by International Myeloma Working Group (IMWG) criteria25,26; had an Eastern Cooperative Oncology Group performance status score of 0–2; had received ≥3 prior lines of therapy, including a PI and an IMiD in any order during the course of treatment, or were double refractory to both a PI and an IMiD; resided in areas where daratumumab was not yet commercially available through local healthcare providers; had not been enrolled in another daratumumab study; and were not eligible for or did not have access to enrollment in another ongoing clinical study of daratumumab. Patients were excluded from the study if they had known chronic obstructive pulmonary disease with a forced expiratory volume in 1 s <50% of predicted normal, known moderate or severe persistent asthma within the past 2 years or current uncontrolled asthma, or clinically significant cardiac disease. Other exclusion criteria were absolute neutrophil count ≤0.5 × 109/L, hemoglobin level ≤7 g/dL (≤4 mmol/L), platelet count <50 × 109/L, alanine aminotransferase level ≥2.5 × upper limit of normal (ULN), total bilirubin level ≥2 × ULN, creatinine clearance ≤20 mL/min/1.73 m2, potassium level <3.0 mEq/L, or corrected serum calcium level >14.0 mg/dL (>3.5 mmol/L).
Study design and treatmentMMY3010 is a multicenter open-label EAP for daratumumab monotherapy. The protocol and amendments were approved by an independent ethics committee or institutional review board, and the study was conducted according to the ethical principles that originated in the Declaration of Helsinki, current International Conference on Harmonization and Good Clinical Practice guidelines, and applicable regulatory and country-specific requirements. All patients provided written informed consent.
Daratumumab (16 mg/kg) was administered intravenously (IV) every week for 8 weeks (Cycles 1–2), every 2 weeks for 16 weeks (Cycles 3–6), and every 4 weeks thereafter in 28-day cycles until disease progression, loss of clinical benefit, unacceptable toxicity, or study conclusion (enrollment closure upon receipt of health authority approval of daratumumab in Brazil) or if daratumumab became available with reimbursement. To reduce the incidence of infusion-related reactions (IRRs), patients received the following pre-infusion medications approximately 1 h before daratumumab infusion: acetaminophen (650−1000 mg IV or orally), an antihistamine (diphenhydramine 25−50 mg IV or orally, or equivalent), and methylprednisolone (100 mg IV for the first and second infusion, 60 mg IV for subsequent infusions). For the prevention of delayed IRRs, all patients received corticosteroid orally (20 mg of methylprednisolone or equivalent) on the 2 days following all daratumumab infusions. For patients with a higher risk of respiratory complications, the following post-infusion medications were considered: an antihistamine (diphenhydramine 25−50 mg or equivalent), a short-acting β2-adrenergic receptor agonist (eg, salbutamol aerosol), and control medications for lung disease (eg, inhaled corticosteroids with or without long-acting β2-adrenergic receptor agonists for patients with asthma; long-acting bronchodilators such as tiotropium or salbutamol with or without inhaled corticosteroids for patients with chronic obstructive pulmonary disease).
Assessments and statistical analysesSafety evaluations included monitoring treatment-emergent adverse events (TEAEs) that were serious adverse events (SAEs), grade ≥3 TEAEs, and TEAEs of special interest (bronchospasm and IRRs). The incidence, severity, and type of TEAEs, as well as the relationship of TEAEs to the study drug and any action taken in response to TEAEs, were reported.
The analysis population included all patients who received at least 1 dose of daratumumab. Unless otherwise specified, continuous endpoints were summarized using descriptive statistics; categorical endpoints were summarized using frequencies and percentages. Treatment exposure and reasons for discontinuation of study treatment were tabulated. Investigator-assessed best disease responses were based on IMWG criteria.27 The Kaplan–Meier method was used to analyze PFS based on investigator-assessed disease progression.
ResultsPatient demographics and baseline characteristicsBetween 13 September 2016 and 9 May 2017, a total of 49 patients were enrolled at 6 study sites in Brazil and received ≥1 dose of daratumumab. Patient demographics and baseline characteristics are shown in Table 1. Median age was 65.0 years, approximately half (53.1%) of the patients were male, and all patients received ≥3 prior lines of therapy.
Patient Demographics and Baseline Characteristics.
| N = 49 | |
|---|---|
| Age, y | |
| Median (range) | 65.0 (50−81) |
| Category, n (%) | |
| 18-<65 | 24 (49.0) |
| 65-<75 | 21 (42.9) |
| ≥75 | 4 (8.2) |
| Sex, n (%) | |
| Male | 26 (53.1) |
| Female | 23 (46.9) |
| Race, n (%) | |
| White | 26 (53.1) |
| Black or African American | 5 (10.2) |
| Asian | 1 (2.0) |
| Other | 6 (12.2) |
| Unknown | 7 (14.3) |
| Not reported | 4 (8.2) |
| Baseline ECOG PS score, n (%) | |
| N | 48 |
| 0 | 19 (39.6) |
| 1 | 25 (52.1) |
| 2 | 4 (8.3) |
| Creatinine clearance, mL/min | |
| N | 47 |
| Median (range) | 68.2 (18.4−146.2) |
| Category, n (%) | |
| ≥90 | 12 (25.5) |
| 60-<90 | 15 (31.9) |
| 30-<60 | 14 (29.8) |
| 15-<30 | 6 (12.8) |
| <15 | 0 (0.0) |
| Hemoglobin, g/L | |
| N | 46 |
| Median (range) | 104.8 (63.0−161.0) |
| Category, n (%) | |
| <80 | 6 (13.0) |
| 80−100 | 12 (26.1) |
| >100 | 28 (60.9) |
| Platelet count, 109/L | |
| N | 46 |
| Median (range) | 59.0 (0−387.0) |
| Category, n (%) | |
| <75 | 24 (52.2) |
| ≥75 | 22 (47.8) |
ECOG PS: Eastern Cooperative Oncology Group performance status.
The median (range) duration of follow-up was 11.1 (0.6–17.5) months. At the time of the analysis, 31 (63.3%) patients had discontinued study treatment. The most common reasons for treatment discontinuation were progressive disease (47.0%), adverse event (AE; 6.1%), loss to follow-up, and patient withdrawal (4.1% each).
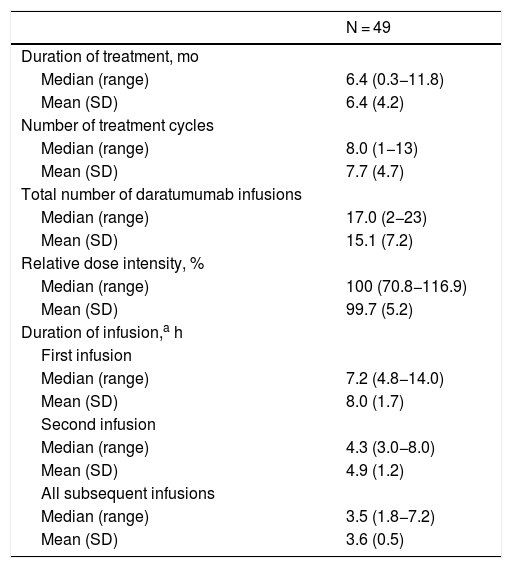
The median (range) duration of treatment was 6.4 (0.3–11.8) months, and patients received a median (range) of 8 (1−13) treatment cycles (Table 2). The median relative dose intensity was 100%. The median duration of daratumumab infusions decreased from the first (7.2 h) to second infusion (4.3 h) and all subsequent infusions (3.5 h).
Treatment Exposure and Infusion Time.
| N = 49 | |
|---|---|
| Duration of treatment, mo | |
| Median (range) | 6.4 (0.3−11.8) |
| Mean (SD) | 6.4 (4.2) |
| Number of treatment cycles | |
| Median (range) | 8.0 (1−13) |
| Mean (SD) | 7.7 (4.7) |
| Total number of daratumumab infusions | |
| Median (range) | 17.0 (2−23) |
| Mean (SD) | 15.1 (7.2) |
| Relative dose intensity, % | |
| Median (range) | 100 (70.8−116.9) |
| Mean (SD) | 99.7 (5.2) |
| Duration of infusion,a h | |
| First infusion | |
| Median (range) | 7.2 (4.8−14.0) |
| Mean (SD) | 8.0 (1.7) |
| Second infusion | |
| Median (range) | 4.3 (3.0−8.0) |
| Mean (SD) | 4.9 (1.2) |
| All subsequent infusions | |
| Median (range) | 3.5 (1.8−7.2) |
| Mean (SD) | 3.6 (0.5) |
SD: standard deviation.
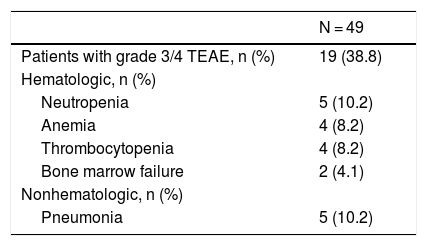
Grade 3 or 4 TEAEs were reported in 19 (38.8%) patients (Table 3). The most frequently reported grade 3 or 4 TEAEs (≥5% of patients) were neutropenia and pneumonia (10.2% each) followed by anemia and thrombocytopenia (8.2% each). Seven (14.3%) patients discontinued therapy due to TEAEs; 3 (6.1%) patients discontinued therapy due to TEAEs that were deemed related to daratumumab and included bone marrow failure, deep vein thrombosis, febrile neutropenia, hypotension, neutropenia, sepsis, and shock (n = 1 [2.0%] each). Seven (14.3%) patients had a TEAE with a fatal outcome; 2 (4.1%) patients experienced a TEAE with a fatal outcome that was deemed related to daratumumab. Of the 2 patients with daratumumab-related TEAEs with fatal outcome, 1 patient had a fatal AE of bone marrow failure, which was within 30 days of the last dose of daratumumab, and 1 patient had a fatal outcome from AEs of febrile neutropenia, deep vein thrombosis, hepatic failure, sepsis, and shock, all of which were within 30 days of the last dose of daratumumab.
Most Common Grade 3 or 4 TEAEs.
| N = 49 | |
|---|---|
| Patients with grade 3/4 TEAE, n (%) | 19 (38.8) |
| Hematologic, n (%) | |
| Neutropenia | 5 (10.2) |
| Anemia | 4 (8.2) |
| Thrombocytopenia | 4 (8.2) |
| Bone marrow failure | 2 (4.1) |
| Nonhematologic, n (%) | |
| Pneumonia | 5 (10.2) |
Shown are grade 3 or 4 TEAEs occurring in ≥2 patients.
TEAE: treatment-emergent adverse event.
SAEs were reported in 19 (38.8%) patients, with 14 (28.6%) patients experiencing grade 3 or 4 SAEs. The most common (occurring in ≥2 patients) any-grade SAEs were pneumonia (10.2%), bone marrow failure, febrile neutropenia, chest pain, deep vein thrombosis, hypertension, and dyspnea (4.1% each). The most common (≥2 patients) grade 3 or 4 SAEs were pneumonia (10.2%) and bone marrow failure (4.1%).
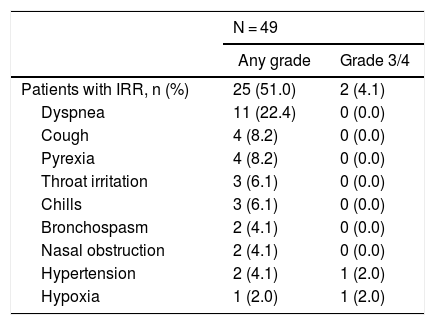
IRRs were reported in 25 (51.0%) patients and were primarily grade 1 or 2 in severity (Table 4). The most frequently reported IRRs (≥5% of patients) were dyspnea (22.4%), cough and pyrexia (8.2% each), and throat irritation and chills (6.1% each). Two (4.1%) patients experienced grade 3 or 4 IRRs (hypoxia and hypertension). The majority of IRRs occurred during the first infusion (23 patients), with no IRRs reported during the second infusion and only 2 patients with IRRs during subsequent infusions.
Most Common Any-Grade and Grade 3 or 4 IRRs.
| N = 49 | ||
|---|---|---|
| Any grade | Grade 3/4 | |
| Patients with IRR, n (%) | 25 (51.0) | 2 (4.1) |
| Dyspnea | 11 (22.4) | 0 (0.0) |
| Cough | 4 (8.2) | 0 (0.0) |
| Pyrexia | 4 (8.2) | 0 (0.0) |
| Throat irritation | 3 (6.1) | 0 (0.0) |
| Chills | 3 (6.1) | 0 (0.0) |
| Bronchospasm | 2 (4.1) | 0 (0.0) |
| Nasal obstruction | 2 (4.1) | 0 (0.0) |
| Hypertension | 2 (4.1) | 1 (2.0) |
| Hypoxia | 1 (2.0) | 1 (2.0) |
Shown are any-grade IRRs occurring in ≥2 patients and/or grade 3 or 4 IRRs occurring in ≥1 patient.
IRR: infusion-related reaction.
An investigator-assessed best response of partial response or better was achieved by 20 (40.8%) patients, including 11 (22.4%) patients with a partial response and 8 (16.3%) patients with a very good partial response; 1 (2.0%) patient achieved a complete response on day 113 of daratumumab treatment (Figure 1). Minimal response was reported in 2 (4.1%) patients, and stable disease was observed in 9 (18.4%) patients. Overall clinical benefit was seen in 31 (63.3%) patients.

Investigator-assessed ORR in patients with RRMM from Brazil.
Note: Response percentages do not add up to ORR due to rounding.
CR: complete response; ORR: overall response rate; PR: partial response; RRMM: relapsed and/or refractory multiple myeloma; VGPR: very good partial response.
The median investigator-assessed PFS was 8.25 (95% confidence interval [CI], 5.55–17.54) months (Figure 2). The 12-month estimated PFS rate was 41.7% (95% CI, 26.9–55.9).
Discussion
The purpose of this EAP was to provide early access to daratumumab treatment while the medication was not commercially available and to collect additional safety data. The data from this EAP for patients with RRMM from Brazil further support the acceptable safety profile of daratumumab monotherapy in patients with MM who have received at least 3 prior lines of therapy, including a PI and an IMiD, or who are refractory to both a PI and an IMiD. The safety results from the current study are consistent with those observed in the phase 1/2 GEN501 study and the phase 2 SIRIUS study,14–16 and no new safety concerns were observed. A small number of patients discontinued treatment due to daratumumab-related TEAEs, and none of these TEAEs were reported in more than 1 patient. Similar to the daratumumab monotherapy clinical studies, IRRs were reported in approximately half the patients, were primarily grade 1 or 2 in severity, and most occurred during the first infusion. Median durations of daratumumab infusions were also very similar to those observed in previous monotherapy trials,22 with infusion durations decreasing with successive infusions. The safety results for the Brazilian cohort of the EAP are also comparable to those observed for other EAP patient cohorts from the United States, Spain, Italy, Russia, and the United Kingdom.23,24 While patients from the Brazilian cohort were treated for a longer duration (median 6.4 months) than patients in the United States cohort (median 1.9 months) or the combined cohort of patients enrolled from Spain, Italy, Russia, and the United Kingdom (median 4.2 months), the incidence of grade 3 or 4 TEAEs (39% vs 50% vs 60%, respectively) and SAEs (39% vs 35% vs 48%, respectively) was comparable or lower for patients from Brazil.
Although not a primary endpoint of the study, efficacy was also evaluated as part of the EAP. For the Brazilian cohort reported here, the overall response rate was approximately 41%, which is higher than the response rate reported in the combined analysis of the GEN501 and SIRIUS clinical studies (31%),16 the EAP United States cohort (23%),23 and the combined cohort of patients enrolled from Spain, Italy, Russia, and the United Kingdom (33%).24 Of note, the majority (84%) of patients in the pooled GEN501 and SIRIUS population were refractory to lenalidomide.16 The median PFS (8.25 months) was also comparable to that observed in the combined analysis of GEN501 and SIRIUS (4.0 months)16 and in the European and Russian EAP patient cohorts (4.63 months).24
In addition to its demonstrated safety and efficacy as monotherapy in patients with heavily pretreated RRMM, daratumumab has also shown significant clinical benefits in combination with standard-of-care regimens across the MM disease spectrum. In phase 3 clinical studies (POLLUX, CASTOR, ALCYONE, and MAIA), the addition of daratumumab to standard-of-care regimens reduced the risk of disease progression or death by at least 44%, nearly doubled the rates of complete response or better, and induced a ≥3-fold increase in minimal residual disease (MRD)–negativity rates at a 10–5 sensitivity threshold versus standard of care alone in patients with RRMM and transplant-ineligible NDMM.17,18,20,21 In Part 1 of the phase 3 CASSIOPEIA study, the addition of daratumumab to bortezomib/thalidomide/dexamethasone as pretransplantation induction and posttransplantation consolidation therapy significantly increased the proportion of patients achieving deep responses, including MRD negativity, and reduced the risk of disease progression or death by 53% compared with bortezomib/thalidomide/dexamethasone alone.19 Part 2 of CASSIOPEIA is currently underway and is evaluating the effect of maintenance treatment with daratumumab monotherapy versus observation.
ConclusionData from this EAP for patients from Brazil demonstrated a safety and efficacy profile for daratumumab monotherapy that was consistent with that observed in clinical studies of daratumumab monotherapy in patients with heavily pretreated RRMM. No new safety concerns were identified. These results further confirm the safety profile of daratumumab in patients with RRMM.
FundingThis study (ClinicalTrials.gov identifier: NCT02477891) was funded by Janssen Research & Development, LLC, which provided support for collection, analysis, and interpretation of data; Janssen employee-authors were involved in the decision to submit the article for publication. Editorial and medical writing support were provided by J. Matthew Kuczmarski, PhD, of MedErgy and were funded by Janssen Global Services, LLC.
Conflicts of interestEdvan de Queiroz Crusoé received research funding from Janssen and received fees from Janssen, Amgen, Celgene, and Takeda. Huiling Pei and Damila Trufelli are employees of Janssen and hold stock in Johnson & Johnson. Mariana Fernandez is an employee of Janssen. Flávia Cristina Fernandes Pimenta, Angelo Maiolino, Nelson Siqueira de Castro, and Luciana Barreto Herriot have no conflicts of interest to disclose.
Data availabilityThe data sharing policy of Janssen Pharmaceutical Companies of Johnson & Johnson is available at https://www.janssen.com/clinical-trials/transparency. As noted on this site, requests for access to the study data can be submitted through Yale Open Data Access (YODA) Project site at http://yoda.yale.edu.
The authors thank all investigators who contributed to this project, the patients who participated in this study, the staff members at the study sites, the data and safety monitoring committee, and the staff members involved in data collection and analyses.
These data were previously presented in part at the 17th International Myeloma Workshop; 12–15 September 2019; Boston, Massachusetts.


