



Autoimmune hemolytic anemia (AIHA) is a rare, life-threatening disease in pediatrics. This article describes the clinical features, diagnostic workup, treatment and outcome in patients with AIHA.
MethodMedical charts of under 18-year-old patients with AIHA treated at a tertiary Brazilian institution from 2006 to 2021 were retrospectively reviewed. Data analysis was primarily descriptive, using medians, interquartile ranges, and categorical variables presented as absolute frequencies.
Main resultsTwenty-four patients (14 female, 10 male) were evaluated in this study. The median age at diagnosis was 5.99 years (range: 0.25–17.1 years) and the median hemoglobin level was 4.85 g/dL (range: 4.17–5.57 g/dL). Most had warm antibodies (83.3 %). Twelve patients (50 %) had known underlining diseases, four (16.6 %) presented with AIHA concomitant with acute infectious diseases and three (12.5 %) had an undetermined post-vaccine association. Steroids and intravenous immunoglobulin were first-line therapy in 23 cases. Seven patients (29.1 %) required second and third-line treatments (rituximab, cyclophosphamide and splenectomy). The median follow-up period was 4.4 years (range: 1.0–6.7 years). Thirteen patients (54.1 %) were discharged, five cases (20.8 %) were lost to follow-up and no patient died. The median age for the six remaining patients was 11.53 years (8.5–14.7) with all of them having complete responses with no further therapies.
ConclusionMost cases of AIHA are secondary to an underlying systemic disease or have a possible correlation with infections/vaccines and respond to steroids. The second and third-line therapies for refractory and relapse cases remain a dilemma. A prospective, multicenter study is essential to address the best therapeutic combinations.
Autoimmune hemolytic anemia (AIHA) is an immunologic disorder characterized by autoantibodies targeting self-antigens on the surface of red blood cells (RBC), with or without complement involvement. The annual incidence is approximately 0.81/100,000 in under 18-year-old patients.1–3
AIHA can be classified as primary (with no associated underlying diseases) or secondary (associated with a disease process, transplants, drugs, or vaccines).1,4,5
Laboratory diagnosis is based on the detection and identification of RBC autoantibodies. According to the isotype and thermal characteristics, AIHA is classified as warm (w-AIHA), cold (cold agglutinin disease - CAD, cold agglutinin syndrome - CAS, paroxysmal cold hemoglobinuria - PCH), and mixed (Table 1). Warm AIHA is the most common type, accounting for 60–70 % of all cases in pediatrics.6
Classification of autoimmune hemolytic anemia.
AIHA: autoimmune hemolytic anemia; DAT: direct antiglobulin test; Ig: immunoglobulin.
(Adapted from “Diagnosis and management of newly diagnosed childhood autoimmune haemolytic anaemia.
Recommendations from the Red Cell Study Group of the Paediatric Haemato-Oncology Italian Association”2.
The term CAD is currently used for chronic AIHA typically seen in adults with chronic lymphoproliferative disorders. CAS refers to cold AIHA in children. It accounts for 20–25 % of all AIHA cases and most commonly occurs with acute infections (Epstein-Barr virus - EBV and Mycoplasma pneumoniae). Other agents have also been associated such as cytomegalovirus, influenza, varicella, hepatitis C, rubella, mumps and measles.2,4,7 Rarely, CAS occurs as a primary process. Mixed-AIHA is extremely rare representing less than 5 % of all cases.4 AIHA is a clinically heterogeneous disease that can represent a potentially fatal condition.1,5
In pediatric AIHA, the therapeutic recommendations are mostly based on retrospective studies, case reports or treatment of adult populations. The management depends on the severity and the speed of anemia development, the type of antibody involved and whether the AIHA is primary or secondary.4
The first-line pharmacotherapy for w-AIHA is corticosteroids. Approximately 50–80 % of patients are corticosteroid-responsive.8 Intravenous immunoglobulin (IVIg) has been used as adjunctive therapy in more severe cases with an efficacy of 54.5 %.1,2,9
The indications for second-line therapy, such as rituximab, are based on no response to first-line treatment, early relapse and steroid-intolerance.1,2
Immunosuppressants (cyclophosphamide, azathioprine, mycophenolate mofetil, cyclosporine and sirolimus) are currently considered third-line treatments.4,9
Splenectomy is recommended for patients who do not have any response or for those who suffer relapse after rituximab therapy.10
This article describes the clinical features, diagnostic workup, treatment and outcomes in children and adolescents diagnosed with AIHA at a tertiary institution over the past 15 years.
Material and methodsAfter obtaining Institutional Review Board approval (59,056,922.2.0000.0068) and written informed consent from parents and eligible patients, a retrospective chart review was performed of all pediatric patients with AIHA seen at a tertiary public hospital in São Paulo, Brazil from 2006 to 2021.
The electronic medical records were searched for under 18-year-old patients with a diagnosis of AIHA (codes 59.1 and 59 of the International Classification of Diseases version 10).
The eligibility criteria were AIHA (hemoglobin - Hb below two standard deviation scores) with features of hemolysis (low haptoglobin level and/or elevated lactate dehydrogenase and/or unconjugated hyperbilirubinemia and/or reticulocytosis) and a positive direct antiglobulin test (DAT). Anemia was defined as severe if Hb level <7 g/dL.8 Based on the isotype and thermal properties of the autoantibody, the AIHA was classified as w-AIHA, CAS, PCH, or mixed-AIHA.
The exclusion criteria were an isolated positive DAT without anemia, Evans syndrome and presence of schizocytes in the blood smear with a negative DAT.
Primary and Secondary AIHA were analyzed. In primary AIHA, no associated underlying condition was identified as a trigger. Secondary AIHA was associated with autoimmune diseases (AID), lymphoproliferative diseases, solid tumors, primary immunodeficiencies (PID) identified by whole exome sequencing, systemic lupus erythematosus (SLE - based on the American College of Rheumatology criteria 2019), chronic infections (EBV, human immunodeficiency virus - HIV, Helicobacter pylori, cytomegalovirus and hepatitis C virus), documented acute viral or bacterial infections at the time of diagnosis, post-transplant AIHA following unrelated bone marrow (HSCT) or solid organ transplantation, hemoglobinopathy-related AIHA, post-vaccine and drug-induced hemolytic anemia.5,9,11,12
Data collected included age, sex, race, ethnicity, comorbidity, signs/symptoms, laboratory results, treatment, and outcomes.
Complete response (CR) to treatment was defined by normal Hb level for age without transfusion or features of hemolysis. Patients with Hb <8 g/dL were considered to show no response (NR). Partial response (PR) was defined by Hb levels between the normal Hb level for age and 8 g/dL.7 Duration of response was calculated from the time treatment was administered until the last day of follow-up for those who remained in complete or PR.
The analysis of data was primarily descriptive. Continuous variables are reported as medians and interquartile range (1st and 3rd quartiles). Categorical variables are presented in absolute frequencies.
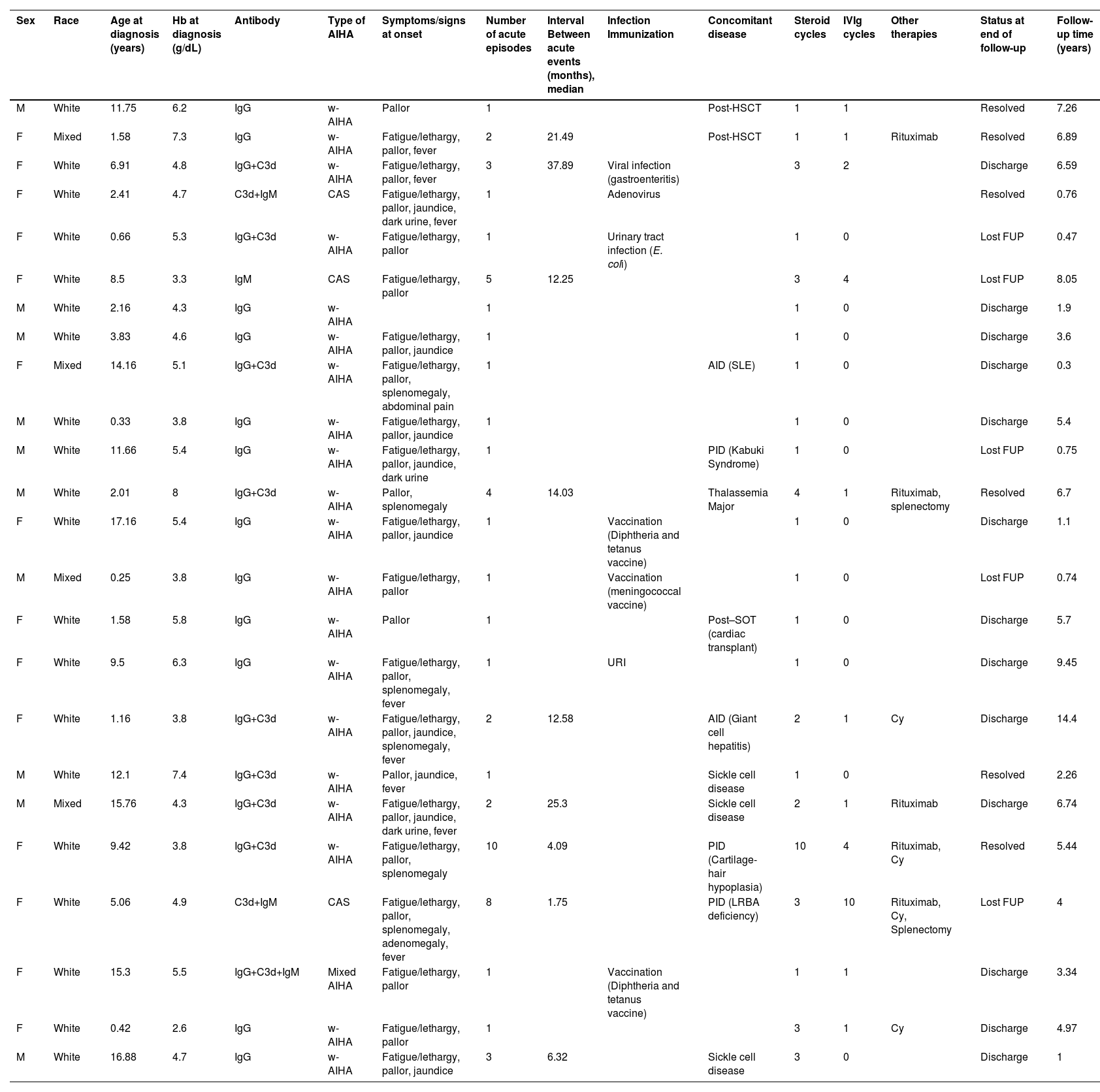
ResultsTwenty-four patients were evaluated in this study. The sex ratio was 1.4F:1.0 M (14 females to 10 males). The median age at diagnosis was 5.99 years (range: 0.25–17.1 years). The clinical and laboratory characteristics of patients are presented in Table 2.
Patients’ clinical and laboratory characteristics.
w-AIHA: Warm autoimmune hemolytic anemia; CAS: cold agglutinin syndrome; Hb: hemoglobin; IVIg: intravenous immunoglobulin; FUP: follow-up; URI: Upper respiratory infection; SLE: Systemic lupus erythematosus; PID: Primary immunodeficiency disorder; AID: autoimmune disease; Post-HSCT: Post-hematopoietic stem cell transplantation; Post-SOT: Post-solid organ transplant; Cy: Cyclophosphamide.
The median Hb level at diagnosis was 4.85 g/dL (range: 4.17–5.57 g/dL) with the anemia being severe in 21/24 (87.5 %) patients. Twenty patients (83.3 %) presented w-AIHA with the DAT type being consistent with IgG in 12 cases (50 %) or IgG plus complement (C3d) in eight patients (33.3 %). Three patients (12.5 %) presented with CAS and one (4.1 %) presented with mixed AIHA. Pallor (95.8 %), fatigue or lethargy (79.1 %), jaundice (37.5 %), and fever (33.3 %) were the most common symptoms. Eight patients (33.3 %) presented with splenomegaly.
Until the last clinic visit, AIHA was considered primary in five patients (20.8 %). Twelve patients (50 %) had secondary AIHA due to a known underlying disease: hemoglobinopathies (n = 4), AID (n = 2), PID (n = 3), post-HSCT (n = 2) and post-heart transplant (n = 1). Four cases were concomitant with acute infections: gastroenteritis with no identified agent (n = 1); upper respiratory infection (URI) caused by adenovirus (n = 1); Escherichia coli urinary tract infection (n = 1) and URI with no viral identification (n = 1; Table 2).
Three patients had an undetermined post-vaccine association according to the World Health Organization causality assessment of an adverse event following immunization.13 The reported vaccines were diphtheria-pertussis-tetanus (DPT: n = 2) and meningococcal (n = 1). The median time between the vaccination and the first symptoms was 8.5 days.
Consanguinity was reported in one patient (4.1 %) with PID. No patient had an AID linked to first-degree relatives.
Treatment analysis according to AIHA etiology indicated that, of the patients who were diagnosed with primary disease (n = 5), three (60 %) received monotherapy (methylprednisolone 30 mg/kg/d × 3 days, followed by oral prednisone 1–2 mg/kg/d for at least 21 days), one (20 %) was treated with steroids and IVIg: 1 g/kg/day × 2 days), and one needed to receive second-line therapy with cyclophosphamide (2–3 cycles of 400–1000 mg/m2 IV every three weeks - Figure 1).
 CR: Complete response; PR: Partial response; IVIg, intravenous immunoglobulin; Cy, cyclophosphamide.")
Patients who possibly had AIHA secondary to a known underlining disease (n = 12) were treated with corticosteroid monotherapy (n = 5), steroids, and IVIg (n = 1) or second-line therapy: rituximab (n = 2), cyclophosphamide (n = 1), rituximab plus splenectomy (n = 1), rituximab plus cyclophosphamide (n = 1), rituximab plus cyclophosphamide and splenectomy (n = 1).
The simultaneous management of AIHA and acute infection identified one spontaneous hematological recovery 11 days after the diagnosis of adenovirus URI. Two cases received corticosteroid monotherapy and one underwent steroid plus IVIg treatment.
Patients who had recently been vaccinated had a full hematological response after corticosteroid monotherapy (n = 2) or steroids plus IVIg (n = 1). No second-line therapy was necessary (Table 2).
Overall, the patients experienced a median of two exacerbation episodes (range: 1.0–2.25) with the median time between acute events being 12.58 months (range: 6.32–21.49 months).
The median follow-up period from the diagnosis was 4.4 years (range: 1.0–6.7 years). In thirteen cases, the follow-up was discontinued because the patient was considered cured (54.1 %); five patients (20.8 %) were lost to follow-up and no patient died.
At the time of the last follow-up, the median age for the six remaining patients was 11.53 years (range: 8.5–14.7 years). All cases were in CR with no need of further treatment.
Four patients (17.4 %) developed systemic arterial hypertension and one (4.3 %) presented Pneumocystis carinii infection secondary to immunosuppression.
DiscussionFrom 2007 to 2022, six case series of AIHA in under18-year-olds were published in the literature.11,14,16–18 The largest prospective study (166 patients) was from the French national pediatric network.14 Contrary to the unexplained tendency for male predominance reported in three studies, 11,14,15 most of the current patients were female (1.4:1 ratio).
According to previous reports, the distribution of age at diagnosis varies significantly (16 months to 11 years).11,14–17 In the current study, the median age was 5.99 years and the median Hb level was 4.85 g/dL, lower than reported in other studies.11,14–17 In concordance with the review of Soochow University, 15 the under one-year-old patients of this study tended to present with a lower Hb level at diagnosis (median Hb: 3.8 g/dL). The presentation was severe in 87.5 %, corroborating the potential lethal condition.
The exacerbation rate of acute episodes varies between 22.8–35.6 % however the median number of episodes and interval between acute exacerbations were not described in previous studies.11,15–17 A median of two acute episodes was observed in 37.5 % of these patients and the median interval between exacerbations was 12.58 months.
Similar to the French database, secondary AIHA comprises about 50 % of the current cohort.10,14 In early childhood, AIHA is generally acute and transient, mainly associated with viral and bacterial infections. In teenagers, there is an increased association with an underlying systemic illness.11
Similar to previous studies, 11,14 17 % of these patients presented infections at the time of the diagnosis. AIHA generally develops in patients within 1–2 weeks of the onset of infections, mostly CAS.7 The incidence of vaccine-related AIHA is unknown. Although three of the current cases (12.5 %) presented AIHA after vaccination, no proven association could be determined.
AIHA reflects a state of immune dysregulation and may show or precede AID or PID several years before disease diagnosis.19 AID and PID are observed in up to 17 % and 13 % of AIHA cases, respectively.8,12,14,15,17 In the current study, five patients (20.8 %) had these etiologies (AID: 8.3 %; PID: 12.5 %). Therefore, in addition to investigating rheumatological diseases, it is essential to provide a thorough immunologic and genetic workup at the clinical onset and a careful follow-up.
Four patients (16.6 %) presented secondary w-AIHA associated with hemoglobinopathies under chronic transfusion therapy. Thalassemia syndromes and sickle cell disease may be complicated by AIHA; these conditions are rare with a prevalence from 1.0 to 6.4 %.5,20 Regarding triggers, previous splenectomy, recent transfusions, alloimmunization, infections and pregnancy are associated with the development of anti-RBC autoimmunity.20 The mechanisms that can lead to autoimmunity include the modification of RBC membrane antigens, exposure to foreign antigens, molecular mimicry after infections and the spread of hidden epitopes during the hemolytic process.5
The incidence of AIHA in allogeneic HSCT recipients ranges between 1.5–2.4 %. Most patients develop AIHA during the first year after the first HSCT.21 In this study two patients (8.3 %) developed w-AIHA, on average, 4.25 months post-HSCT. The immunological dysregulation may be due to the use of calcineurin inhibitors or anti-thymocyte globulin administered before unrelated donor HSCTs, infections, graft-versus-host disease, transfer of relatively naïve T lymphocytes and mismatched donor cells, and delayed immune reconstitution.22
One case (4.1 %) presented AIHA one year after heart transplantation. Autoimmune cytopenia has been reported in 6.9 % of post-heart transplant patients with the most common type being AIHA (76.9 %). It may be associated with infections, ABO incompatibility and immune dysregulation. Heart transplant patients usually require higher levels of immunosuppression, which may contribute to autoimmunity.23
Regarding the therapeutic response, in this study, eighteen patients (78.3 %) had CR following first-line therapies similar to the literature (steroid: 78 %; steroid plus IVIg: 22 %), in a median recovery time of 23.8 days (range: 18–34 days) from the treatment.8
One patient (4.1 %) presented mixed-AIHA and was treated as w-AIHA. Mixed-AIHA is often characterized by lower hemoglobin levels and a worse prognosis than w-AIHA. Two or more lines of therapy are frequently needed.7,25 The patient in the current study reached CR following first-line treatment (corticosteroids).
Seven patients (30.4 %) required second-line treatments and 71.4 % received rituximab (375 mg/m2 weekly x 4). The response rate was 80 %, which is similar to what is reported in previous studies, in a median recovery time of 6.29 weeks (range: 5.71–7.0 weeks).4,25 Despite possible complications (hypogammaglobulinemia, hepatitis B virus reactivation and, rarely, progressive multifocal leukoencephalopathy) rituximab may be preferred to splenectomy due to the lower risk of infective complications.4,6,7 Two of the current patients (8.3 %) were submitted to splenectomy with one of them relapsing 5.92 months after the procedure. Previous publications indicate that nearly one-third of the cases relapse after splenectomy, mostly within 1–2 months. A better response occurs in those without underlying AID or hematology malignancy.10
Four patients (16.6 %) received 2–3 cycles of IV cyclophosphamide (400–1000 mg/m2 every three weeks) as third-line therapy with a response rate of 50 %, similar to that reported in the literature.4
Recommendations to treat CAS have been based on theoretical considerations, case reports, and expert opinion. Transfusion, when indicated, can be considered safe as compatibility problems are usually not encountered in CAS. Non-pharmacological management consists of thermal protection.24 No therapy has been established for CAS associated with malignancies except for treating the underlying disease. In bacterial infection-associated CAS, antibiotics should be instituted. In the current study, one patient presented spontaneous hematological recovery (adenovirus-associated CAS), one patient presented primary CAS responsive to corticosteroids and one case had severe refractory CAS responsive to rituximab and splenectomy. This patient had the diagnosis of PID (lipopolysaccharide-responsive beige-like anchor - LRBA deficiency) 34 months after the treatment.24
Plasma exchange is an option for patients with fulminant hemolysis unresponsive to RBC transfusion. Response to complement inhibition has been described in single cases but there is no systematic study on complement inhibition.4,25
Finally, HSCT may represent the possibility of definitive treatment for AIHA. This option has been reported but success is limited and should be reserved for patients with severe refractory AIHA and life-threatening disease for whom all other therapies have failed.4,25,9
Although this analysis has limitations related to the retrospective design, small sample size, loss to follow-up and single-center involvement, it describes the heterogeneity of the clinical manifestation and highlights the different etiologies of a life-threatening disease in pediatrics. Most cases were associated with underlying systemic illnesses. A minority of patients received second and third-line therapies, the definition of which remains a dilemma. A prospective, multicenter study with an adequate number of cases and rigorous control to avoid loss to follow-up is essential to understand this severe immune condition and to address the best therapeutic combinations.



