




Chimeric Antigen Receptor (CAR) T cells have tremendous potentials for cancer treatment; however, various challenges impede their universal use. These restrictions include the poor function of T cells in tumor microenvironments, the shortage of tumor-specific antigens and, finally, the high cost and time-consuming process, as well as the poor scalability of the method. Creative gene-editing tools have addressed each of these limitations and introduced next generation products for cell therapy. The clustered regularly interspaced short palindromic repeats-associated endonuclease 9 (CRISPR/Cas9) system has triggered a revolution in biology fields, as it has a great capacity for genetic manipulation.
MethodIn this review, we considered the latest development of CRISPR/Cas9 methods for the chimeric antigen receptor T cell (CAR T)-based immunotherapy.
ResultsThe ability of the CRISPR/Cas9 system to generate the universal CAR T cells and also potent T cells that are persistent against exhaustion and inhibition was explored. Conclusion: We explained CRISPR delivery methods, as well as addressing safety concerns related to the use of the CRISPR/Cas9 system and their potential solutions.
The increasing risk of cancer in human society is a major issue, so finding safe and effective treatments has become one of the leading aims of researchers around the world.1 A kind of immunotherapy that uses the body's own immune system has become a new and promising treatment for cancer. It offers more effective and lasting treatment than conventional therapies, such as chemotherapy, radiotherapy and surgery.1,2 The T-cell therapy is a type of immunotherapy that enables the body's own T cells to raid the cancer. In the body's lymphatic tissues, hundreds of billions of T cells live and circulate in the bloodstream, detecting and killing infected cells.3 T cells react to infected cells via T cell receptors (TCRs). They detect the antigen presented by major histocompatibility complex (MHC) molecules on infected cells and antigen-presenting cells (APCs).4 The T-cell transfer therapy, also called adoptive immunotherapy/adoptive cell therapy/immune cell therapy/tumor-infiltrating lymphocyte (TIL) therapy/and gene-modified T-cell therapy, includes two main types: engineered TCR therapy and chimeric antigen receptor T cell (CAR T) therapy. Both methods involve collecting the body's own immune cells, reactivating and expanding high numbers of them in the laboratory and then returning the cells via a needle into the vein.5 This method of immune cell therapy, which has revolutionized the meaning of drugs, uses genetically altered autologous/allogeneic T cells as a drug to revitalize a patient's own immune system to destroy cancer cells.4,6 In contrast to monoclonal antibody drugs, which only bind specifically to their targets to trigger the immune system, T cells have the ability to proliferate and directly destroy cancer cells. In addition, T cells have an immunological memory and said memory of anti-cancer capacity can be stored in the patient's body for a longer period of time than with traditional medicines.2
CAR T cells as genetically modified T cellsTCRs recognize intracellular antigens via the human leukocyte antigen (HLA1)-dependent procedure. The HLA-I, a heterodimer protein, exists in the membranes of almost all of the nuclear cells and presents small endogenous peptides, such as tumor antigens to the immune system.7 T cells must acquire some characteristics before they can reach their own full potential, including the creation of a specific connection between the TCR against the tumor antigen, as well as the activation and proliferation of tumor antigen-specific T cells. T cells must also be able to be placed in diseased areas and to overcome any immunosuppressive agents in the way and release their lytic compounds and cytokines.8
Tumor-specific T cells cannot be obtained from most patients with cancer, except in relatively rare cases. However, T cells that respond to tumors can be genetically altered, which involves inserting some genes into them that encode cell surface receptors and are capable of detecting tumor-associated antigens TAAs.9 These receptors can be chimeric antigen receptors (CARs), which are synthesized using molecular biology techniques. These artificial T cell receptors give T cells a new capacity for targeting a specific protein. The receptors are named chimeric because they mix both the antigen-binding function and T-cell activation duty into a single receptor.10
CAR constructionThe molecular structure of the CAR depends on the target antigen and the generation of CAR T cells. Targets usually are surface antigens owning specific epitopes to cancer cells. Having unique targets for cancer cells is essential to avoid unexpected consequences related to autoimmune diseases. These engineered membrane antigens can trigger T cell activation through a mechanism involving antibody-like recognition.11 The CARs contain three major components: an intracellular activation domain for the T cell, a transmembrane hinge domain and an extracellular antigen-recognition domain. The antigen-recognition portion usually consists of a single-chain variable fragment (scFv) taken from hypervariable fragments of an antibody's light and heavy chains (Figure 1). The transmembrane part is taken from the CD28 or CD8. The intracellular part is a signaling domain for the activation of T cells and is usually taken from the cytoplasmic part of the CD3ζ chain of the TCR complex. This domain is often incorporated with the CD27, CD28, CD134 and CD137, that are co-stimulatory molecules that assist in the T cell signaling pathway and, as a result, in turn, induce the proliferation and permanence of the T cell.12
 an extracellular binding element; 2) a transmembrane portion (TM), and; 3) the intracellular signaling domain. Commonly, the binding portion consists of an scFv domain taken from a TAA-specific monoclonal antibody and a signaling domain or domains derived from the TCR activating and co-stimulatory elements.")
The basic structure of the CAR. CARs consist of three segments: 1) an extracellular binding element; 2) a transmembrane portion (TM), and; 3) the intracellular signaling domain. Commonly, the binding portion consists of an scFv domain taken from a TAA-specific monoclonal antibody and a signaling domain or domains derived from the TCR activating and co-stimulatory elements.
The first-generation CARs contain only the CD3ζ signaling part providing just signal 1 for the T cell activation. However, both signal 1 and signal 2, produced by costimulatory molecules, are essential for the effective activation of T cells. Primary clinical trials on the first-generation CAR T cell for cancer treatment display very restricted responses and the CAR-engineered cell lasted at low levels for only a few weeks or months. This suggests that first-generation CAR T cells are lacking enough activation signals needed for T cell long-term proliferation and efficient anti-tumor effects. Second-generation CARs were produced by inserting an additional signaling portion in the intracellular part of a CAR to enable further activation signals in the T cell.13 This portion is taken from a co-stimulation molecule, such as the 4-1BB, CD28, CD134 CD70, CD80 and CD86. The CD28 enables frequent antigen stimulation, as well as the triggering of the proliferation of T cells. The CD28, an Ig superfamily member, was the first costimulatory molecule applied in second-generation CAR T cells and is more popular than other costimulatory molecules. However, in most FDA-approved CAR-T cell products, the 4-1BB, a TNF-R superfamily member that supports CAR-T cell persistence, has been utilized. The CD27 stimulates the T cell proliferation, but there is also some speculation that the CD27 plays a role in the formation of the T cell immune memory. The CD134 and CD137 are similar in their roles, as they are both costimulatory molecules, as well as being both members of the tumor necrosis factor (TNF) receptor family. The CD70, CD80, CD86 and CD137 have been demonstrated to elevate the proliferation of T cells, in addition to the release of cytokines, once motivated via the antigen recognition.2,14 Third-generation CARs have been generated via inserting multiple co-stimulatory portions in intracellular domains of the CAR structure. In the fourth generation CAR T cells, also called TRUK (T cell redirected for universal cytokines killing), more genetic manipulations have been developed, enabling the expression of proliferative T cell ligands that can simultaneously stimulate the 4-1 BBL or pro-inflammatory cytokines (IL2).15 The fourth generation of CAR T cells, immediately after antigen recognition on tumor cells, begins to secrete large amounts of perforins, enzymes and tumor necrosis agents, leading to the apoptosis of tumor cells. Compared to the previous three generations, TRUK cells have more advantages in acting on immunosuppressive cells around the tumor and, therefore, have a higher destructive power. The fifth, or next, generation CAR T cells, with better proliferation and persistence, constructed based on the second generation, in which TCR alpha and beta chains were ablated and a fragment of the IL-2 receptor β (IL-2Rβ) was added to the co-stimulatory domain, instead of the OX-40/CD27, to induce cytokine signaling.16 The generations of CAR T cells are summarized in Figure 2.
Candidate targets for CAR
The selection of an appropriate target for the CAR receptor is a very important determinant and needs a comprehensive assessment. Complexes of the MHC/antigen that are tumor-specific are considered desirable targets for T cell-based therapy. Tumor antigens that are CAR targets have traditionally been mostly limited to cell surface peptides.17 Not only proteins, but also post-translational modification molecules, such as carbohydrate or even glycolipid molecules, could be the potential targets.18 The basic elements for an optimal target antigen include “specificity”, “functional dependence” and minimum “antigen escape”. Therefore, the ideal target should express uniformly on tumor cells, express through all stages of the tumor and, importantly, have restricted expression in other tissues to decrease “off-tumor” side effects.19 Antigens are classified based on their expression manners that include the tumor-specific antigen (TSA), a TAA, as well as the cancer germline antigen (CGA). To achieve substantial tumor elimination, the majority of tumor cells should be targeted by CAR-T cells, meaning that the selected target antigen should sufficiently cover the tumor cells.18 Currently, most of the targets used in CART-based therapies that are clinically effective well comply with the criteria identified above. For instance, the CD19 is a good target for B cell malignancies, the CD20 antigen is used for B-cell lymphoma and the CD22 is suitable for the B cell maturation antigen (BCMAB) and acute B lymphoblastic leukemia B-ALL.17 The CD30 that is a member of the TNF receptor (TNFR) superfamily is efficient in Hodgkin lymphoma. In addition, there are some other targets with high coverage that deserve further verification, such as the C type lectin-like molecule-1 (CLL-1) for acute myeloid leukemia blasts. Other hopeful targets to treat hematological malignancies that have been generated and are under clinical trial include the CD33 and CD123 to treat acute myeloid leukemia (AML), the CD133 for acute lymphocytic leukemia (ALL) and AML, the B-cell maturation antigen (BMCA) and CD138 for multiple myeloma (MM), inactive tyrosine-protein kinase transmembrane receptor (ROR1) for ALL and chronic lymphocytic leukemia (CLL) and the immunoglobulin Kappa chain (Igκ) for CLL.6,17
Limitations and strategies to overcome using the CRISPR/Cas9 systemUp to now in CAR T clinical trials, the main origin of the T cells has been autologous, or those originating from the patients themselves. However, autologous T cell-based therapies face limitations. First, since the product must be generated from each patient's cells, it is a time-consuming and costly process that can delay the availability of treatment, especially in patients with highly proliferative diseases. The second barrier is the low quantity and quality of primary autologous T cells in patients receiving chemotherapy or radiotherapy. In addition, the heterogeneous expression of tumor antigens, as well as the escape mechanisms of tumor cells in the immune system, necessitate the use of CAR T cells that can target multiple tumor antigens simultaneously. Moreover, the immunosuppressive tumor microenvironment (TME) creates barriers to T cell activity and induces the T cells to differentiate and exhaust. In addition to limitations regarding the anti-tumor effect of these cells, the most prominent limitations due to the use of these cells is their side effects, two of the most important of which include Cytokine release syndrome (CRS) and neurotoxicity.
Using allogeneic T cells derived from healthy donors as ‘‘universal’’ CAR T cells can overcome the limitations and produce large amounts of fully functional cells.20 Despite many favorable features, allogeneic T cells also face challenges. First, the recognition of recipient cell alloantigens by allogeneic T cells can cause intensive graft-vs-host disease (GVHD). Second, the recognition of exogenous HLA molecules on donor T cells may result in rapid allorejection. Therefore, both the HLA and TCR must be silenced or deactivated in allogeneic universal CAR T cells.20
To produce the highest quality universal CAR T cells, a highly efficient gene editing tool is needed to alter multiple genes simultaneously, involving minimum manipulation to produce final products with high qualities. The clustered regularly interspaced short palindromic repeats (CRISPR) method is a simple and highly accurate gene editing method that is highly versatile and has the unique potency to easily edit multiple genes. In human primary T cells, the clustered regularly interspaced short palindromic repeats-associated endonuclease 9 (CRISPR/Cas9) gene editing method makes it possible to simultaneously knock out several gene loci with very high efficiency.21
The CRISPR/Cas9 systemThe CRISPR/Cas9 technology derives from the type II immune system of bacteria and archaea that protects them against invading elements, such as viruses (named bacteriophages), plasmids and any other foreign nucleic acids.22 Upon activating the system, short fragments release from invading foreign DNA and insert themselves between the repeat sequences of CRISPR arrays within the prokaryotic host genome. The arrays include “protospacers”, short fragments of DNA, matching part of the corresponding invading DNA. These elements develop a “memory” in the bacterium or archaeon that, when an invader with the same or similar sequence is encountered subsequently, permits the host to release an RNA section from its CRISPR arrays, targeting foreign DNA for destruction.23 In this struggle, transcripts from the CRISPR repeat arrays are processed into CRISPR RNAs (crRNAs), which hybridize with a second trans-activating CRISPR RNA (tracrRNA). The crRNA/tracrRNA hybrid associates with the bacterial endonuclease called Cas9 (CRISPR-associated protein 9) and guides it to a complementary target DNA of invading viral. The Cas9 cleaves the genome, provided that it be adjacent to a short sequence named protospacer adjacent motif (PAM).24 The CRISPR-based immunity creates the basis of CRISPR-Cas9 technology, which can be used to alter genes within other organisms. This elegant and creative defense system has been present in diverse aspects of medicine.21
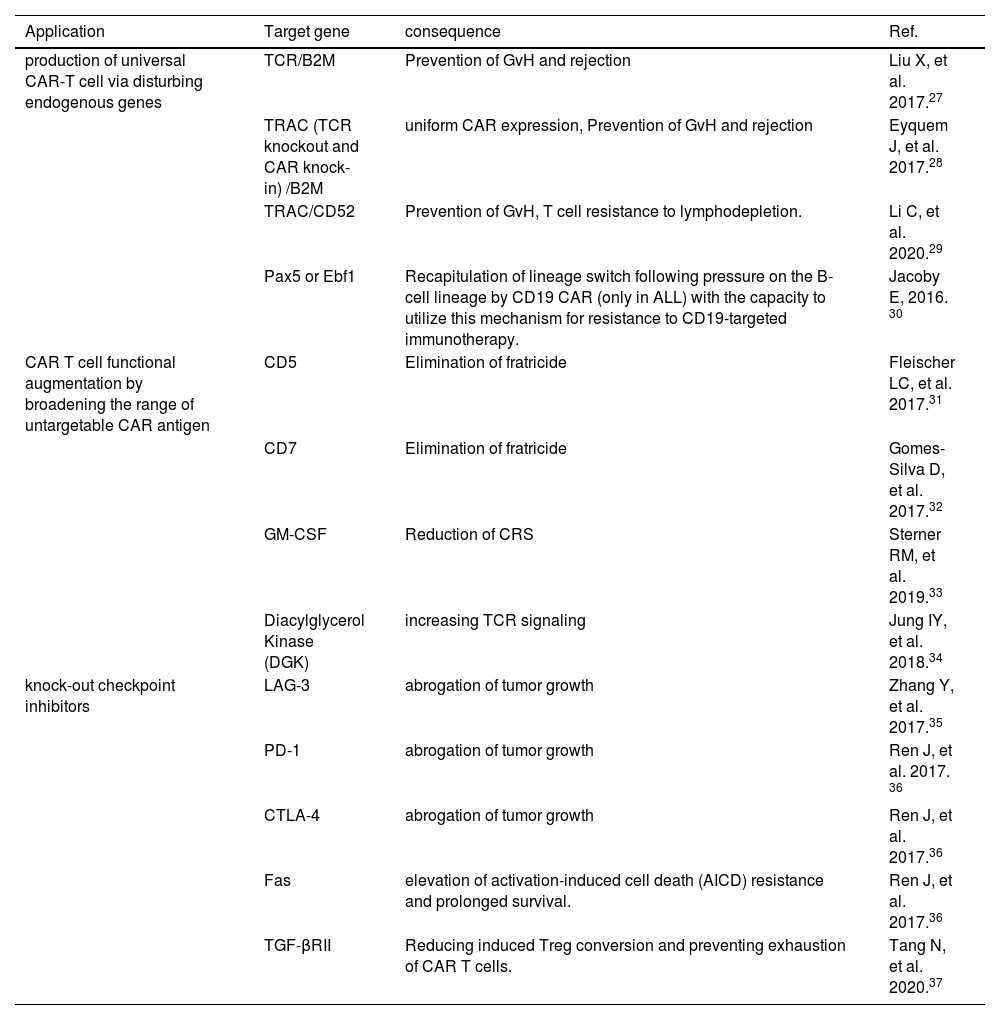
How CRISPR improves the CAR T cell capabilityThe CRISPR/Cas9 genomic editing provides unlimited potential to create next-generation T cell products to fight cancers and other diseases. The CRISPR/Cas9 can be used to edit the TRAC locus of a CAR in order to generate universal CAR T cells.25 Using the CRISPR/Cas9, upgraded allogeneic universal CAR T cells that are defective simultaneously in two (TCR beta chain, beta-2-microglobulin (B2M)) or three genes (TRAC, B2M, PD-1) have been generated. These CAR T cells, without causing any GVHD, keep their function both in vitro and in vivo. In addition, further disruption of the TCR, PD-1 and HLA class I-negative CAR T cells resulted in remarkably improved in vivo anti-cancer activities.26 The application aspects of the CRISPR/Cas9 can be summarized in three major classes: production of universal CAR-T cell via disturbing endogenous MHC and TCR molecules to destroy graft-vs-host (GvH) and host-vs-graft (HvG), CAR-T augmentation by broadening the range of untargetable CAR antigen and knock-out checkpoint inhibitors for improving antitumor ability (Figure 3) (Table 1).
 the production of the universal CAR-T cell via disturbing endogenous MHC and TCR molecules to destroy GvH and HvG; 2) CAR-T augmentation by broadening the range of untargetable CAR antigen, and; 3) knock-out checkpoint inhibitors for improving antitumor ability.")
Improvement of CAR-T cell therapy by CRISPR/Cas9 technology: three major aspects of the CRISPR/Cas9 application in the CAR-T cell therapy: 1) the production of the universal CAR-T cell via disturbing endogenous MHC and TCR molecules to destroy GvH and HvG; 2) CAR-T augmentation by broadening the range of untargetable CAR antigen, and; 3) knock-out checkpoint inhibitors for improving antitumor ability.
Applications of the CRISPR/Cas9 system for improving CAR T cells.
| Application | Target gene | consequence | Ref. |
|---|---|---|---|
| production of universal CAR-T cell via disturbing endogenous genes | TCR/B2M | Prevention of GvH and rejection | Liu X, et al. 2017.27 |
| TRAC (TCR knockout and CAR knock-in) /B2M | uniform CAR expression, Prevention of GvH and rejection | Eyquem J, et al. 2017.28 | |
| TRAC/CD52 | Prevention of GvH, T cell resistance to lymphodepletion. | Li C, et al. 2020.29 | |
| Pax5 or Ebf1 | Recapitulation of lineage switch following pressure on the B-cell lineage by CD19 CAR (only in ALL) with the capacity to utilize this mechanism for resistance to CD19-targeted immunotherapy. | Jacoby E, 2016. 30 | |
| CAR T cell functional augmentation by broadening the range of untargetable CAR antigen | CD5 | Elimination of fratricide | Fleischer LC, et al. 2017.31 |
| CD7 | Elimination of fratricide | Gomes-Silva D, et al. 2017.32 | |
| GM-CSF | Reduction of CRS | Sterner RM, et al. 2019.33 | |
| Diacylglycerol Kinase (DGK) | increasing TCR signaling | Jung IY, et al. 2018.34 | |
| knock-out checkpoint inhibitors | LAG-3 | abrogation of tumor growth | Zhang Y, et al. 2017.35 |
| PD-1 | abrogation of tumor growth | Ren J, et al. 2017. 36 | |
| CTLA-4 | abrogation of tumor growth | Ren J, et al. 2017.36 | |
| Fas | elevation of activation-induced cell death (AICD) resistance and prolonged survival. | Ren J, et al. 2017.36 | |
| TGF-βRII | Reducing induced Treg conversion and preventing exhaustion of CAR T cells. | Tang N, et al. 2020.37 |
Strategies that promote the function of T cells are beneficial for immunotherapy. Immune checkpoint regulators, such as cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1), are negative regulators of T cell activity.38 The activity of engineered T cells is also inhibited by immune checkpoint regulators that have opened a new insight for the immunotherapy of cancer. However, the up-regulation of immune checkpoint regulators or their inhibitory ligands (CTLA4 and PDL1) may be a limiting factor for the CAR T cell perdurability and function, that will lead to unwanted clinical outcomes. Using the CRISPR/Cas9 technology to ablate immune-checkpoint regulators improves the efficiency of T cell-based immunotherapy, first applied by Su et al., for knocking out PD-1 in CAR T cells and resulted in the enhancing of the cytotoxicity the CAR T cell, without affecting the T cell viability. Since then, various research groups have applied different methods and presented similar results.39,40
CAR T cell functionality augmentation via the CRISPR/CasSeveral studies have shown that using the CRISPR/cas9 increases the CAR T activity. It has been reported that knocking out the Granulocyte-Macrophage Colony Stimulating Factor (GM-CSF) gene by the CRISPR/cas9 not only increases the CAR T cell antitumor activity and survival, but also decreases the neuroinflammation and the CRS.26 It is known that the CAR T cell targeting of the CD7 can destroy both by targeting existing CD7 markers on themselves, an action termed fratricidal activity. Silva et al. indicated that knocking out CD7 in CAR T cells using the CRISPR/Cas9 prevents fratricidal activity followed by CAR T cell immunotherapies.41 Moreover, knocking out the CD7 and TRAC in CAR T cells increased the efficacy in the treatment of T cell acute lymphoblastic leukemia (T ALL).42 Jung et al. reported that using the CRISPR/Cas9 to knock out the diacylglycerol kinase (DGK), an enzyme that metabolizes the diacylglycerol gene, causes an increase in the CD3 signaling and an improvement in the T cell function via the enhancement of the TCR signaling.43
Safety concerns related to the CRISPRImmune-related CRISPR/cas9 side effects lead to some restrictions in its therapeutic applications, which are summarized in the following and some solutions are listed. The delivery system of the Cas protein and single guide RNA (sgRNA) can recall either the host's innate or acquired immune system. Furthermore, the secondary structure of the sgRNA might be recognized by pattern diagnosis receptors and elicit an immune response.44 Thus, further studies are needed to verify clinical restrictions of these immune reactions.38 The CRISPR/Cas9 system makes double-strand breaks in the DNA that may induce the p53-mediated apoptosis in receptor cells and decrease the cell viability. Therefore, p53 inhibitors can improve the efficacy of the CRISPR-mediated gene editing, so the p53 function should be monitored to reduce the risk of p53 mutation in cells.45,46 Some unpredicted large mutagenesis-like deletions and genomic rearrangements may also occur at the CRISPR target site, on-target mutation that may have pathogenic effects.47 For a solution, the drug-dependent apoptosis can be induced by inserting the suicide receptor gene next to the CRISPR platform. These receptors bind to a drug through their extracellular portion and use a caspase9 protein endodomain as an effector.48 In the CAR T cell therapy, using such a suicide gene strategy reduces the risks of the CRISPR-mediated gene editing and increases the safety of cells in case of unpredicted effects.49 However, the main restricting concern in clinical trials is off-target effects (OTEs) of genome editing.50 Strategies in reducing OTEs include respectively: the designing of more appropriate guide RNA (gRNA), choice of more specific endonucleases and lessening of the exposure time to endonucleases. Appropriate software tools for the gRNA designing, such as azimuth, benching and elevation, that predict the risk of off-target slicing, can remarkably decrease OTEs.51 To improve the specificity of the gRNA, chemical modification of the Crispr RNS (crRNA) to interrupt its pairing to off-target sequences and creating truncated gRNAs at 5′ are some of the approaches.52,53 The elevated specificity can be obtained in another popular way that is the precise choice of the appropriate endonuclease. The Francisella novicida Cas9 (FnCas9) and Cpf1 have been reported to be more specific than the SpCas9.54,55 Moreover, changing some amino acid residues of the SpCas9 has led to engineered Cas9 proteins, with high fidelity and low OTEs, such as the eSpCas9, HypaCas9for and SpCas9-HF1. These modified Cas9 proteins might change their mechanism of action due to changes in the binding strength of the target DNA, or a change in the nuclease activation model (HNH domain).56,57 Another kind of nuclease was created by fusing two subunits, the dead cas9 (dCas9), whose nuclease domain is inactivated, and the FOK1 cleavage domain. This chimeric protein must be dimerized to make breaks in the DNA. Since two close sequences are recognized by this dimer enzyme and two distinct designed sgRNAs, this approach increases specificity.58 The Cas9 nickase variant (nCas9) uses a similar strategy and is mutated in one of its two nuclease domains. Each nCas9 is guided with a separate sgRNA and cleaves only one strand of DNA, thereby resulting in increased specificity.59 The third main strategy for reducing OTEs is to limit the exposure time to CRISPR nuclease, which also reduces on-target efficacy.51 One strategy to reduce the time of Cas9 exposure is to use delivery systems, such as ribonucleoprotein (RNP) complexes, Cas9 mRNA and integrase-deficient lentiviral vector.60,61 However, despite the many benefits of the RNP delivery system, its unpleasant immune effects should also be considered. Other strategies to low-off Cas9 exposure time include: (i) controlling the Cas9 expression by using the doxycycline-inducible promoter that leads to a regulated expression; (ii) regulating the Cas9 activity via cell-printable compounds and creating Cas9-degron, Cas9-intein and split-Cas9, and; (iii) designing a self-destructive construct consisting of a Cas9 protein that targets its own gRNA system.62,63
DeliveryDepending on ex vivo or in vivo trials, various delivery systems and several CRISPR formats (nucleic acid, protein or ribonucleoprotein) might be used.64 For the ex vivo delivery of the CRISPR/Cas9 in nucleic acid and RNP formats, some routine methods, such as the electroporation or microinjection, as well as newfound techniques, such as transmembrane internalization assisted by membrane filtration (TRIAMF),65 or induced transduction by osmocytosis and propane betaine (iTOP) have been used.66 For in vivo usage, because of mentioned safety problems, such as immunological responses and off-target mutations, the selection of the delivery method should be made more carefully. Up to now, the most frequently used delivery techniques apply viral methods for transducing cells, which also have some limitations. They include unwanted immune activation (adenoviral vectors), increased rate of off-target mutations caused by the long expression of Cas9, creating insertional mutagenesis (mostly in lentiviral vectors) and restrictions related to viruses packaging (adeno-associated viral vectors).67 Because of the restrictions of viral-based delivery methods, non-viral delivery techniques, such as lipid, polymeric and inorganic particles have been developed. An additional superiority of these synthetic particles is their potency for delivering the CRISPR/Cas9 as a ribonucleoprotein complex. Successful treatment of the Duchene muscular dystrophy has been obtained in a mouse model by this system, in which gold nanoparticles have been used. Compared to other synthetic carriers, gold nanoparticles are non-toxic and, therefore, preferred.68,69
ConclusionThe CRISPR/Cas9 gene-altering technology has presented hopeful applications and explorations for creating the next-generation CAR T cells. Some examples are the universal CAR T cells by defecting endogenous TCR and HLA, more powerful CAR T cells by disrupting inhibitory modulators, controllable CAR T cells containing suicide genes or inducible safety switches and novel CAR T cells avoiding self-killing by knocking-out of the targeted antigens.70 However, the specificity and efficiency of the CRISPR/Cas9 gene-editing technology are associated with some concerns. The first issue is the off-target effects introducing random mutations that activate oncogenes or impact tumor-suppressor genes resulting in unwanted deleterious consequences. Various solutions, such as the accurate selection of the target position and optimized methods for designing the sgRNA and Cas9 activity, have been provided to minimize the risks of off-target effects.71,72 The efficient and nontoxic delivery of the CRISPR system into CAR T cells is another challenge and some main delivery methods with simplicity, safety and flexibility have been created. Thus, the CRISPR/Cas9 technology, with technical development for decreasing off-target effects and modifying delivery efficiency, provides a powerful potential to create novel CAR T cells products to fight cancers and other diseases.
Since these technologies are growing in scope and potency, ethical and monitoring guidelines should also be thoughtfully made to guarantee a balance between mankind's usage benefits of the enormous potential and the risk of their misuse. After all, the lack of relevant clinical studies could be considered as an essential restraining cause for its large-scale clinical use because of the mentioned concerns and possibly costly manufacturing procedure.
FundingThis research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Ethical approvalNot Applicable.
Data statementData sharing is not applicable to this article, as no new data were created or analyzed in this study
Notes on contributorsConceptualization and Supervision: Elham Roshandel.
Writing, review & editing: Maryam Vahdat Lasemi, Maryam Mehravar, Abbas Hajifathali.
Writing of the original draft: Afshin Mohammad Alizadeh, Mohammad reza Moshari.
All authors revised the manuscript and approved the final paper, conception or design of the study.
The authors would like to thank the staff of the Hematopoietic Stem Cell Research Center, Shahid Beheshti University of Medical Sciences, Tehran, Iran, for providing the possibility of writing this manuscript.


