

Hematopoietic progenitor cell (HPC) transplantation is the only curative treatment for patients with sickle cell disease (SCD). More than one thousand patients with SCD have undergone this treatment so far.1 Usually, the preferred source of HPC for SCD patients is the bone marrow, as transplant of mobilized HPC has been associated with a higher rate of graft-versus-host disease.2 Despite this, many patients with SCD are transplanted with mobilized HPC. However, most of the donors for SCD patients are their siblings, who have a high probability of presenting sickle cell trait (SCT). There was concern as to whether SCT donors were at a higher risk of presenting adverse effects, such as vaso-occlusive crisis, due to HPC mobilization with granulocyte colony-stimulating factor (G-CSF). To our knowledge, there are only three articles that address this issue and none of them has shown a higher incidence or severity of adverse reactions in SCT donors. Furthermore, efficacy of mobilization was not inferior to non-SCT donors.3–5
In Brazil, the regulation recently promulgated by the agency that regulates blood transfusion and cell therapies, the Agência Nacional de Vigilância Sanitária (ANVISA), prohibits HPC mobilization in SCT donors, which limits treatment access for patients with SCD. There are many situations in which peripheral blood would be the preferred source for HPC, as it usually provides a much higher HPC content than bone marrow, which is an important advantage associated with this source. Herein we report three cases out of 43 transplants for SCD with safe and successful HPC mobilization and harvesting in SCT donors whose products were transplanted into their siblings.
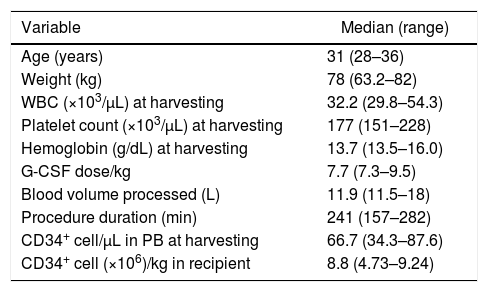
Two male donors, aged 28 and 31, and one female donor, aged 36, underwent HPC mobilization with G-CSF, at doses of 7.3μg/kg/day to 9.5μg/kg/day for four days, with harvesting being carried out on Day 5. All donors mobilized well (Table 1) and CD34+ cell yields were adequate. Leukapheresis procedures were performed using COBE SPECTRA equipment in two cases and a SPECTRA OPTIA device (TERUMOBCT, Lakewood, CO, USA) in one. HPC harvesting required just one session in all three cases. Donors had no unexpected adverse reactions such as pain crises suggestive of vaso-occlusion during mobilization or harvesting. Two recipients engrafted on Days 14 and 17 and are still alive with donor hematopoiesis three and four years after transplant, without graft-vs.-host disease (GVHD). The other recipient died of infection five months after transplant with no sign of engraftment. This patient had lost engraftment of a previous bone marrow transplantation and had had chronic GVHD restricted to the skin, which was in remission at the time of the infusion of HPC from peripheral blood.
Characteristics of hematopoietic progenitor cell mobilization with granulocyte colony-stimulating factor (G-CSF) in three sickle cell trait donors and CD34+ cell yield.
| Variable | Median (range) |
|---|---|
| Age (years) | 31 (28–36) |
| Weight (kg) | 78 (63.2–82) |
| WBC (×103/μL) at harvesting | 32.2 (29.8–54.3) |
| Platelet count (×103/μL) at harvesting | 177 (151–228) |
| Hemoglobin (g/dL) at harvesting | 13.7 (13.5–16.0) |
| G-CSF dose/kg | 7.7 (7.3–9.5) |
| Blood volume processed (L) | 11.9 (11.5–18) |
| Procedure duration (min) | 241 (157–282) |
| CD34+ cell/μL in PB at harvesting | 66.7 (34.3–87.6) |
| CD34+ cell (×106)/kg in recipient | 8.8 (4.73–9.24) |
WBC: white blood cell count; PB: peripheral blood.
SCT is considered a benign state and individuals with this mutation are generally asymptomatic.6 However, SCT has been associated with renal disorders and vaso-occlusive crises in specific situations, such as exposure to extremely high altitudes. What is more, there have been some reports of severe complications, such as multi-organ dysfunction in sickle cell disease patients treated with G-CSF.4,7 Because of this, there was some concern, and there still is to some degree, that the administration of G-CSF, a hematopoietic growth factor with pro-inflammatory effects, to SCT donors of HPC could provoke sickling or other complications. Nevertheless, this was not observed in the three articles published so far. The first one demonstrated that HPC mobilization with filgrastim and the processing of large blood volumes by aphaeresis procedures in eight SCT donors appeared to be safe.3 More recently, Al-Khabori et al. reported three cases of successful mobilization and collection of HPC in donors with SCT.4 The largest study on this issue reported 41 successfully mobilized SCT donors of HPC.5 SCT donors presented better mobilization of CD34+ cells than Caucasian donors (123±91/μL vs. 74±46/μL). The rate of adverse events observed in SCT donors was similar to non-SCT donors in all three publications. Herein, we observed the same results as the other authors. The three SCT donors presented very good mobilization of CD34+ cells and cell collection by leukapheresis with no significant complications.
In conclusion, we state, based on the literature and on our own experience, that mobilization of HPC with G-CSF in individuals with SCT and cell harvesting by aphaeresis are safe procedures. Furthermore, there are other mobilizing agents, such as plerixafor, with little or no pro-inflammatory effects, the use of which would circumvent the pro-inflammatory effects of G-CSF and will probably also be used in healthy HPC donors. ANVISA made a grave mistake by prohibiting the exploration of this type of procedure. Mobilized HPC in SCT donors can be extremely useful for a wide range of patients, especially for those with SCD, who are, as expected, much more likely to have a SCT donor as a member of his or her family.
Conflicts of interestThe authors declare no conflicts of interest.


