


The guidelines project is a joint initiative of the Associação Médica Brasileira and the Conselho Federal de Medicina, aiming to bring information in medicine to standardize management and help decision-making during treatment. The recommendations of this article were elaborated by the Associação Brasileira de Hematologia, Hemoterapia e Terapia Celular (ABHH). The treating physician should evaluate all possible medical approaches, considering the patient's characteristics and clinical status.
This article presents the guidelines on sickle cell disease: primary stroke prevention in children and adolescents.
Description of the method used to collect evidenceThese guidelines result from a systematic evidence-based review centered on the Evidence-Based Medicine movement, where clinical experience is integrated with the ability to critically analyze and apply scientific information rationally, thereby improving the quality of medical care.
The questions were structured using the Patient/Problem, Intervention, Comparison and Outcome (PICO) system, allowing the generation of evidence search strategies in Medline (via PubMed) and using a manual search. The data recovered were critically analyzed using discriminatory instruments (scores) according to the type of evidence. After identifying studies that potentially substantiate the recommendation, the level of evidence and degree of recommendation were calculated using the Oxford Classification.1
BackgroundSickle cell disease (SCD) encompasses a group of autosomal recessive hemoglobinopathies whose genetic mutation leads to the replacement of glutamic acid by valine in position 6 of the beta-globin chain (β6 Glu → Val), resulting in the variant hemoglobin S (HbS → α2β2s). The most common and severe SCD is sickle cell anemia (SCA, homozygous HbS = HbSS). Other forms of SCD are double heterozygosis as HbS/β0-thalassemia (HbSβ0, severity as HbSS), HbS/β+thalassemia, HbSC disease (HbSC), HbSD disease (HbSD), and other associations less frequent. The main clinical manifestations of the disease are related to anemia and vaso-occlusive events caused by HbS polymerization, a process dependent on hypoxia that leads to inflammation, excessive adhesion of erythrocytes to the activated endothelium, ischemia-reperfusion injury, a hypercoagulable state, and dysregulation of vascular tone.2(D)
Stroke is a significant cause of morbidity and mortality in individuals with SCD, leading to severe motor and neurocognitive sequelae.3,4(D) The stroke physiopathology can be explained by vasculopathy and occlusion caused by the sickle erythrocytes with stenosis of cerebral vessels of the circle of Willis (middle cerebral artery, anterior cerebral artery, anterior communicating artery, internal carotid artery, posterior cerebral artery, and posterior communicating artery).5(D) Patients with HbSS develop vasculopathy and occlusion at specific sites as distal internal carotid artery (ICA) and the proximal segments of the middle cerebral artery (MCA) and anterior cerebral artery (ACA).6,7(A) Studies have been conducted to identify concurrent risk factors for cerebrovascular disease, and the detection of high cerebral blood flow velocity (CBFV) in arterial segments of the polygon of Willis by conventional transcranial doppler (TCD) has been confirmed as the major risk factor for the development of ischemic stroke in children and adolescents with SCA.8,9(B) TCD can detect intracranial arterial stenosis caused by arterial vasculopathy associated with SCA.7(A),10(B) TCD is a non-invasive, portable, and relatively inexpensive method that uses specific areas of the skull (acoustic windows) to access the intracranial arterial circulation and to measure CBFV in the polygon of Willis arteries. Unlike angiography, considered the gold standard test, it is less invasive and does not present any risk of complications.
Many advances have been made in treating SCD and in the primary prevention of stroke, such as chronic packed red blood cell (pRBC) transfusions, drugs that induce fetal hemoglobin (HbF), and hematopoietic stem cell transplantation.4(D)
Clinical questions- 1.
Is there evidence that transcranial doppler should be performed in all patients with sickle cell disease for primary stroke prevention?
This question intends to assess the role of TCD or transcranial doppler imaging (TCDI) in diagnosing SCD patients at risk for stroke.
Adams et al. published the first study to establish the accuracy of TCD in assessing the risk of ischemic stroke in asymptomatic children with SCA. TCD and cerebral angiography were compared to determine TCD sensitivity and specificity in detecting intracranial arterial lesions characterized by a reduction in the vascular lumen diameter equal to or greater than 50%. They included 33 patients with SCA , ranging from 2 to 30 years (mean age 12±6 years). In 34 TCD exams, 26 of 29 patients (89.65%) had significant abnormalities, with sensitivity and specificity values of 90% and 100%, respectively. Therefore, TCD is a sensitive and specific method to detect arterial vasculopathy in SCA patients.10(B)
In 1992, a prospective, observational study in 190 patients with SCA , aged 3 to 18 years (mean age 8.9±4.2 years), evaluated the relation between TCD and the occurrence of clinical stroke.6(A) Abnormal TCD velocity was defined as a maximum (max) CBFV greater than or equal to 170 cm/second (cm/s) in the MCA (not including ICA), a definition determined by post hoc analysis to maximize the test's predictive success. After a mean follow-up of 29±17 months (range 4-58 months), 283 TCD were performed. Abnormal TCD was found in 23/190 patients (12.10%), and in 7/23 (30.43%), an ischemic stroke was diagnosed. High CBFV in the MCA (TCD ≥170 cm/s) was associated with stroke (Fisher's exact test - p < 0.00001), and the relative risk of stroke was 44 (95% confidence interval (95%CI), 5.5 to 346). The TCD can identify the risk of stroke in patients with SCA.7(A) The extension study included additional 125 children with SCD (HbSS or HbSβ0). Two limits for high CBFV were studied: 170 and 200 cm/s. Forty percent (10/25) of the patients who had max TCD velocity ≥ 200 cm/s had a stroke, while only 2% (7/290) with max TCD velocity < 200 cm/s had a stroke. The risk of stroke was associated with max TCD velocity ≥ 200 cm/s. This value was then established as an abnormal TCD.11(A) The subsequent STOP trial unequivocally proved that TCD could identify the risk of stroke in patients with SCA.7(A)
The prospective phase III randomized Stroke Prevention Trial in Sickle Cell Disease (STOP) assessed whether children with SCD (HbSS, HbSβ0) and abnormal TCD would have reduced risk of a first stroke if they received periodic transfusions of pRBC to maintain HbS < 30%. The study included 130 patients with SCD with no history of stroke, aged between 2 to 16 years (mean age 8.3±3.3 years), and two abnormal TCD velocities. STOP trial categorized TCD velocities according to mean max CBFV in MCA as normal (< 170 cm/s), conditional (≥ 170 cm/s to < 200 cm/s), and abnormal (≥ 200 cm/s). These values were used in future studies.7(A)
The Hydroxyurea to Prevent Organ Damage in Children With Sickle Cell Anemia (BABY HUG) trial performed TCD in 192 children with SCA, mean age 12.6 months (range 7-17 months) and concluded that more studies are needed to validate max TCD velocities that can predict the risk of stroke in children with SCA under two years of age.12(A)
Comparing TCD and TCDI mean velocities in transcranial arteries in 22 children with SCA between 3-14 years (mean eight years), TCDI velocities were lower than TCD for all vessels : CBFV in the MCA were -9.0%, in ICA -10.8%, in ACA -19.3%, in bifurcation -16.3%, and in basilar arteries -23,1%. Therefore, considering TCDI results, hematologists must be careful to indicate chronic pRBC transfusions.13(B) In another study in 22 children (3-16 years) with SCA, the TCDI velocities were generally lower than TCD velocities for the same segment. The MCA CBFV in the TCDI was lower than TCD both on the right (mean 16 cm/s) and on the left (mean 13 cm/s) (p < 0.001 and p = 0.02, respectively). So, the TCDI result is not the same as TCD.14(B) After studying 53 children with SCA, aged 2 to 17 years, the mean max TCDI velocity measurements were significantly lower than those made with the TCD: 10.9% (p < 0.0001) in the MCA, 12.7% (p = 0.002) in the ACA, 2.2% (p = 0.69) in the posterior cerebral artery (PCA), 21.0% (p < 0.0001) in the distal ICA, and 15.3% (p < 0.0001) at the bifurcation of the distal ICA. These values are 10% lower than those obtained from the TCD in the STOP trial.15(A)
However, some studies suggested that the TCDI and TCD results may be similar. In 66 children with SCD and 84 paired exams (TCD and TCDI) performed on the same day by the same sonographer, max CBFV in the MCA, the distal ICA, and the ACA at TCD and TCDI were not significantly different.16(B) Thirty-seven children with SCA, aged 2 to 12 years (mean 7.8±3.0 years) performed TCD and TCDI at the same session, and the max CBFV of the TCDI was determined with and without correction for the angle of insonation in the arteries. The average angle of insonation in the MCA, ACA, ICA, and PCA was 31°, 44°, 25°, and 29°, respectively. TCDI velocities were 20% lower than TCD velocities (p < 0.05), although they are not different from the angle-corrected TCDI velocities.17(B)
A training program to standardize TCD and TCDI is essential to guarantee the results according to STOP trial and the quality of the exam. Fifty-five practitioners from different specialties from three European hematology clinics performed the TCD in 555 patients with SCD before and after training. This training was completed by 23 (42%) specialists. After training, the TCD and TCDI results were closer between the three centers. The stroke risk was different between the three centers before training (p < 0.001) and improved after training (Fischer's Exact = 6.7, p = 0.305; no treatment = 5.6, p = 0.41, treatment = 13.8, p < 0.001), regardless of using TCD or TCDI.18(A)
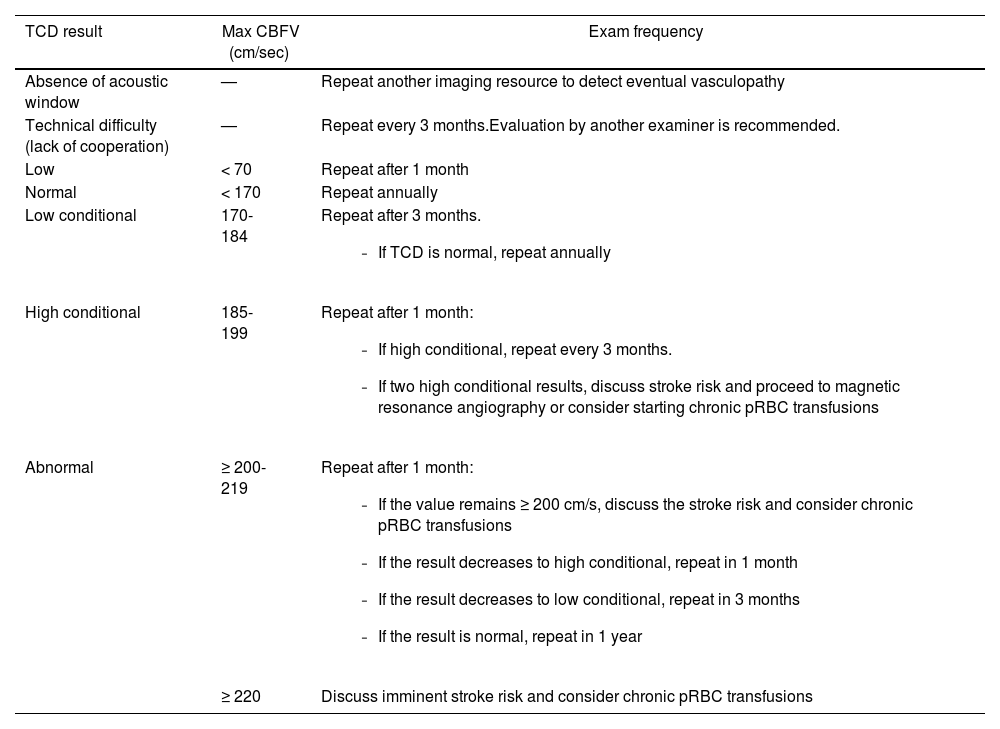
The frequency to perform the TCD recommended by the Brazilian Guidelines for TCD in children and adolescents with SCD depends on the TCD results (Table 1).19(D)
Recommendations for the TCD frequency in SCD patients between 2-16 years old according to the maximum cerebral blood flow velocity in the middle cerebral artery.
Max CBFV, maximum mean cerebral blood flow velocity; pRBC, packed red blood cell; SCD, sickle cell disease (Hb SS and Hb S/ß0); TCD, conventional transcranial doppler.
Adapted from Lobo et al, 2011.19(D)
Recommendations
- 1.
Conventional transcranial doppler (TCD) should be periodically performed in children with sickle cell disease (HbSS/HbSβ0), aged between two and 16 years, by measuring the mean maximum velocity of the middle cerebral artery and internal carotid artery to identify patients at risk for stroke.
- 2.
Considering that transcranial doppler imaging (TCDI) does not have standardized values for indicating red blood cell transfusions, it is recommended that the method used, whether TCD or TCDI, be mentioned in the results report.
- 3.
Training the professional who will perform the TCD is necessary to standardize the exam technique and the reading of the mean maximum velocity of the middle cerebral and internal carotid arteries values.
- 2.
Is there evidence that chronic packed red blood cell transfusions can be used as a treatment to prevent primary stroke in patients with sickle cell disease?
STOP trial was designed to assess the incidence of stroke (ischemic or hemorrhagic) in SCD (HbSS, HbSβ0) patients without previous stroke, but with a high risk to develop it due to abnormal TCD ≥ 200 cm/s), who are on pRBC transfusions regimen to reduce the HbS < 30%. One hundred and thirty children were randomized to standard treatment with folic acid and pRBC transfusions when necessary (n = 67) or pRBC transfusions every three to five weeks (n = 63). There were ten cerebral infarcts and one case of hemorrhagic stroke in the standard group and only one event (ischemic stroke) in the transfusion group (RRA=0.148, 95%CI: 0.036 to 0.177; NNT=6.7, 95%CI: 5.6 to 28). After a mean follow-up of 19.6±6.5 months, the risk of stroke decreases 92% (p < 0.001) in the transfusion group.7(A)
Children between 2-16 years with SCD (HbSS, HbSβ0) at high risk for stroke (TCD ≥ 200 cm/s) who had received chronic pRBC transfusions for at least 30 months and the TCD became normal were randomized to continue with regular pRBC transfusions (n = 38) or to stop transfusions (n = 41). The risk of stroke in the group without regular transfusions was around 45% (p < 0.001). The Optimizing Primary Stroke Prevention in Sickle Cell Anemia (STOP 2) Trial was stopped early after the occurrence of 16 events (14 abnormal TCD and two strokes) among individuals randomized to stop transfusions, and no event was observed in the transfusion group (p < 0.001; RRA=0.390, 95%CI: 0.213 to 0.390; NNT=2.5, 95%CI: 2.5 to 4.6). STOP 2 showed that chronic pRBC transfusions should be continued indefinitely in SCD children with abnormal TCD, since stopping them often increases CBFV to risk levels.20(A) A cohort study with 20-year follow-up in SCA children (mean age 3.7 years, range 1.3-8.3) at risk for stroke (TCD ≥ 200 cm/s) analyzed 92 patients in chronic pRBC transfusions. After a mean follow-up of 6.1 years, no stroke was developed and in 83.5% of patients the TCD normalized.21(B)
The “Post-STOP” trial evaluated the impact of the STOPs Protocols on the incidence of ischemic stroke . Data from 2,808/3,835 (73%) patients who participated in STOP and/or STOP 2 trials and had no stroke were analyzed. After a mean follow-up of 9.1±3.4 years (median 10.3, range 0-15.4), 2.1% (n = 60) patients had the first ischemic stroke with an incidence of 0.24/100 patient-years (95%CI, 0.18 to 0.31). The mean age at a first stroke was 13.7 years (median 13.2, range 3.5 to 28.9). Most strokes occurred in patients in whom the STOP protocol had not been appropriately implemented (63%) due to failure to screen with TCD (38%) and failure in adequate transfusions when TCD was abnormal (25%). About 8% of strokes occurred in children with abnormal TCD who were receiving adequate chronic pRBC transfusions. This failure rate is acceptable as the risk of recurrent stroke was approximately 20% in SCD (HbSS, HbSβ0) patients in regular pRBC transfusions. This trial confirms that chronic pRBC transfusions can reduce ischemic stroke in SCD children and adolescents. The prevention of ischemic stroke requires a complete implementation of the STOP Protocol in treatment centers.22(A)
A meta-analysis confirmed the reduction in stroke (primary prevention) in children and adolescents with SCD (HbSS, HbSβ0) and abnormal TCD velocity (≥ 200 cm/s) receiving regular pRBC transfusions.23(A)
- 3.
Is there evidence that hydroxyurea can be used as a treatment to prevent primary stroke in patients with sickle cell disease?
Hydroxyurea (HU) is a cytostatic and myelosuppressive agent that acts directly on the pathophysiological mechanism of SCD, raising HbF level and improving blood flow through reduced cellular adhesion. The solubility of the cellular hemoglobin becomes stabilized, and the red blood cell (RBC) membrane damage decreases, reducing hemolysis. HU also decreases the RBC-endothelial interactions, reducing expression of RBC, white blood cells, endothelial integrins, and other adhesion molecules, improving microvascular blood flow and reducing pro-inflammatory cell-cell interactions and thrombosis. HU stimulates nitric oxide production, inducing local vasodilation. The HU mechanism of action in SCD reduces symptoms and morbidities of the disease-related vaso-occlusion.24(D)
The STOP trial shown that primary stroke prevention with regular pRBC transfusions is the appropriate treatment for patients with SCD (HbSS and HbSβ0) at high risk of stroke due to abnormal TCD (≥ 200 cm/s).7(A) However, other studies shown the possibility of using HU after a period of pRBC transfusions to reverse TCD to non-critical levels, mainly in cases with a lack of adherence to the transfusions regimen, alloimmunization, and shortages of phenotyped RBC.
A retrospective observational study from the Belgian registry analyzed 127 patients under 19 years with severe SCD treated with HU. During the follow-up period that analyzed 426 patient-years, 3.3 episodes of an acute thoracic syndrome, 1.3 cerebrovascular events, and 1.1 cases of osteonecrosis were observed for every 100 patient-years. In 72 patients evaluated by TCD, 34 were at risk for primary stroke (TCD > 200 cm/s). Reduction in TCD velocities was reported in 11 children, and only one shown a cerebrovascular event (seizures) during a follow-up of 96 patient-years.25(B)
A prospective study including 24 children (mean age: 9.9 years, range 20 months-16 years) with SCA found that the use of HU at the maximum tolerated dose (MTD) for at least six months (range 6-48 months) reduced the TCD velocities by a mean of 13.0 cm/s (95%CI: -20.19, -5.92 cm/s; p = 0.0002). The SCA age-matched control group without HU (n = 24) had a mean increase in TCD velocities, after approximately one year, of +4.73 cm/s (95%CI: -3.24, 12.69). Demographic data are similar between the groups, including the mean initial TCD velocities: 125 cm/s (range 83–206 cm/s) in the treatment group and 128.9 cm/s (range 79–220 cm/s) in the control group. The reduction in TCD velocities in the treatment group was greater than in the control group (p < 0.001).26(B)
In another prospective phase II study, 36 children (mean age 6.8±3.5 years) with SCD who had had TCD greater than or equal to 140 cm/s were treated with HU at MTD. Fifteen children had conditional TCD value (170-199 cm/s) and six had abnormal TCD values (≥ 200 cm/s). Subjects maintained on HU for an average of eight months (10±5) had significant reductions in TCD velocities in the right MCA (166±27 cm/s to 135±27 cm/s, p < 0.001) and in the left MCA (168±26 cm/s to 142±27 cm/s, p < 0.001). Considering the children with conditional TCD, 14 had decreased TCD velocities. The six children with abnormal TCD velocities had decreased it from 216±14 cm/s to 173±31 cm/s (p < 0.001). One new neurologic event occurred in a patient with abnormal TCD after seven months of HU therapy. The overall incidence of new neurologic events was one in 193.2 patient-years or 0.52 events per 100 patient-years.27(B)
The Transcranial Doppler with Transfusions Changing to Hydroxyurea (TWiTCH) trial sought to establish the non-inferiority of HU (alternative arm) in comparison to chronic pRBC transfusions (standard arm) in the primary stroke prevention in children with SCA, TCD ≥ 200 cm/s who received at least 12 months of pRBC transfusions, and no severe vasculopathy at magnetic resonance angiography (MRA). It is a prospective, phase III, randomized, open-label multicenter study in which 121 children aged 4 to 16 years were randomized to continue pRBC therapy (n = 61) or change to alternative therapy with HU at MTD (n = 60). Transfusions in alternative arm were slowly weaned over 4–9 months to protect against stroke during HU dose escalation to MTD. At randomization, the mean TCD velocity in the standard arm was 145±21 cm/s and in the alternative arm was 145±26 cm/s. Children in the standard group maintained HbS <30% and a mean HbF of 25% in the alternative group. The study was discontinued in the first interim analysis as the non-inferiority of HU had been demonstrated (maximum mean TCD velocity of 143±1.6 cm/s vs. 138 cm/s in those receiving pRBC transfusions and HU, respectively, 95%CI: 4.54 (0.10, 8.98), non-inferiority p = 8.82 × 10−16). No child developed stroke, and three in each group developed transient ischemic attack (TIA). However, important information is that all children included in this study had received pRBC transfusions for an average of four years (at least 12 months) before being treated with HU. In the setting of an abnormal TCD, transfusions should always be the initial treatment. The best time to make the transition to HU has not been determined. Any cerebral vasculopathy identified by magnetic resonance imaging contraindicates HU.28(A)
In Nigeria the SCD burden is high and there is an inadequate supply of safe blood. The parents are reluctant to accept regular pRBC transfusions and the treatment cost is self-paid. A prospective observational study was done in 104 children between 2-16 years with SCA and elevated TCD velocities (≥ 170 cm/s) treated with HU at MTD. After a median duration HU therapy of 3.6 years (1 to 8), the mean TCD velocities in 44 patients with abnormal risk (≥ 200 cm/s) decreased from 211.9 (±12.7) cm/s to 180.0 (±21.07) cm/s (p < 0.001), with 29.5% of these patients reaching standard risk (< 170 cm/s). In the high conditional group (185–199 cm/s), the mean TCD velocity decreased from 191.5 (±3.98) cm/s to 165.5 (±20.03) cm/s (p < 0.001) and in the low conditional group (170–184 cm/s) decreased from 179.95 (±2.82) cm/s to 156.80 (±16.27) cm/s (p < 0.001), with 61.7% going to standard risk. Stroke incidence was 0.27/100 patient-years due to one stroke event in a patient with abnormal TCD and no response to HU.29 (A)
Stroke Prevention in Nigeria (SPIN) included 29 children (median age 8.1 years, range 6.1-10.3) with SCA and TCD velocities ≥ 200 cm/s receiving HU moderate fixed-dose (20 mg/kg/day) and 206 children with normal TCD velocities as a comparator group. The median baseline TCD velocity in the HU group was 208 cm/s (range 205-226), and in the comparison group, 134 cm/s (range 118-150), p < 0.001. A decrease in the abnormal TCD was observed after three months of HU therapy (median 183 cm/s) and was sustained after two years (median 163 cm/s), both p < 0.001. Among the comparison group, four developed abnormal TCD measurements after one year and crossed over to the treatment group. There were 19 deaths with no relation to neurologic events and none in HU treatment.30(B)
Considering that standard of care treatment for conditional TCD velocity is observation, the phase III, prospective, randomized Sparing Conversion to Abnormal TCD Elevation (SCATE trial) evaluated the efficacy of HU at MTD versus observation in preventing conversion from conditional to abnormal TCD (≥ 200 cm/s) in 22 children with SCA, median age 5.4 years (range 2.7-9.8). The trial was terminated early because of the slow patient accrual and administrative delay. SCATE showed the cumulative incidence of abnormal conversion as 9% (95%CI: 0-35%) in the HU group and 47% (95%CI: 6-81%) in the observation group arm at 15 months (p = 0.16). Even though, in post hoc analysis, no children on HU and 50% on observation converted to abnormal TCD velocities (p = 0.02). After a mean of 10.1 months, reduction in TCD velocities in patients receiving HU was -15.5 cm/s (95%CI: -32.0 to 1.1), and in the observation group, the TCD velocity had a mean increase 10.2 cm/s (95%CI: -4.8 to 25.2, p = 0.02). No strokes or TIA were related.31(B)
A Brazilian retrospective study in 36 children between two and 16 years with SCD (HbSS or HbSβ+) with elevated TCD velocities (≥ 170 cm/s) also showed the benefits of HU at MTD in reducing TCD velocities, mainly in conditional TCD. The conditional TCD in HU-group had significant velocities reduction (176.8±5.3 cm/s to 162.7±13.9 cm/s, difference of 14.1 cm/s; p = 0.001), data not observed in those without HU (176.3±5.3 cm/s to 170.0±18.6 cm/s, difference of 6.3 cm/s; p = 0.148).32(B)
Another Brazilian retrospective/prospective study with 718 children with HbSS or HbSb0-thalassemia showed 54 children (7.5%, all HbSS, median age at the first TCD 4.9 years) with a high-risk TCD (n = 45) or, when the TCD was inconclusive, with an MRA with severe vascular injury (n = 9). Of these, 51 children started pRBC. Considering the children with high-risk TCD, 29 (67.4%) reverted to low risk and in 18 of them (62%), HU was started at the MTD before transfusions discontinuation. None of these 29 patients had a stroke. Eight children (18.6%) maintained a high-risk TCD, even using the pRBC and HU, and two had a stroke.33(A)
Based on the TWITCH trial, a meta-analysis from the Cochrane Database of Systematic Reviews concluded that switching to HU with phlebotomy is non-inferior to chronic pRBC transfusions in children as primary stroke prevention.23(A) A recent meta-analysis tried to answer the question: Can HU be used in children with SCD and cerebral vasculopathy to prevent chronic complications? Based on two studies (SWITCH and TWITCH), the authors concluded that HU was not superior to chronic pRBC transfusions for reducing neurological events in pediatric patients. They discuss that two studies are too few to show strong recommendations, but, even though, recommended in children with SCD and cerebrovascular abnormality (primary or secondary stroke prevention) chronic pRBC transfusions plus iron chelation therapy instead of HU.34(A)
To primary stroke prevention in patients with SCA, the British Society for Haematology Guideline recommends that children with regular pRBC transfusions for abnormal TCD can be switched to HU if they have received at least one year of regular transfusions and have no MRA-defined severe vasculopathy (level of evidence 1A). Transfusions must be continued until they have reached HU MTD. If TCD velocity is between 170–200 cm/s (conditional risk), they should be treated with HU to prevent progression from conditional to abnormal TCD velocities (level of evidence 1B) at MTD (level of evidence 1C).35(D)
The American Society of Hematology recommendation for children with HbSS or HbSβ0 (ages 2-16 years) who have abnormal TCD velocities and live in a high-income setting is chronic pRBC transfusions for at least a year to reduce the risk of stroke (strong recommendation based on moderate certainty in the evidence about effects). After at least one year of pRBC transfusions and normal MRI and MRA of the brain, even if abnormal TCD velocity, HU at MTD can be suggested to substitute regular pRBC transfusions (conditional recommendation based on low certainty in the evidence about effects). Although, if the children with HbSS or HbSβ0 or compound heterozygous SCD live in low-middle-income settings, the ASH guideline panel suggests HU at least 20 mg/kg/day (fixed dose) or the MTD (conditional recommendation based on low certainty in the evidence about effects).4(D)
Recommendations
- 1.
Conventional transcranial doppler (TCD) must be performed at least once a year and chronic packed red blood cell (pRBC) transfusions should be indicated with abnormal mean TCD velocity (≥ 200 cm/s) as first line treatment to prevent primary stroke in sickle cell disease (HbSS or HbS/β0 thalassemia) patients aged 2 to16 years. After four years (at least 12 months) of pRBC transfusions, normal TCD and magnetic resonance angiography with no severe vasculopathy, hydroxyurea (HU) at maximum tolerated dose (MTD) can be offered to prevent primary stroke, maintaining pRBC transfusions until the patient reach HU MTD.
- 2.
The best time to make transition to HU has not been determined.
- 3.
After transition to HU, TCD doppler must be performed annually and pRBC transfusions restarted if abnormal TCD velocity is detected.
- 4.
For sickle cell disease (HbSS or HbS/β0 thalassemia) patients aged 2-16 years with conditional TCD (170-199 cm/s), HU can be offered to reduce TCD velocities.
- 4.
Is there evidence that hematopoietic stem cell transplantation can be used as treatment to prevent primary stroke in patients with sickle cell disease?
Hematopoietic stem cell transplantation (HSCT) is currently considered the only curative treatment for SCD, being accepted as a safe and effective therapy. Since the first HSCT in SCD patient performed in 1984, several studies have shown positive results, especially when using hematopoietic stem cells from matched sibling donors.36,37(B) Currently, more than 1,250 patients with SCD have been transplanted worldwide. Part of these data was published in 2017 by Gluckman et al., demonstrating excellent overall survival (OS) and event-free survival (EFS) rates, respectively 95% and 93%.36(B)
Recently, Bernaudin et al. published better results in patients under 30 years who received myeloablative conditioning for HSCT. In this study, the 5-year EFS was 97.9% (95%CI, 95.5–100%), confirming a cure rate of 95% after 2000.38(B)
Since these publications, HSCT using bone marrow as stem cell source from a matched related donor and myeloablative conditioning has been considered standard treatment for SCD patients with indication for HSCT. In Brazil, according to ordinance number 1,321, of December 21, 2015, allogeneic HSCT using related bone marrow or cord blood as a stem cell source and myeloablative conditioning is indicated for several clinical situations in SCD patients, as the presence of the previous stroke, clinically significant neurological events, or neurological deficit lasting more than 24 h.
Although several studies have demonstrated the benefit of HSCT in primary stroke prevention, only one article, recently published, explored this issue in the study design since all other studies included patients with and without previous stroke.
Bernaudin et al. presented the results of the first prospective study comparing HSCT with related donors versus standard treatment with chronic pRBC transfusions in patients with SCA and abnormal TCD. Sixty-seven children (median age 7.6 years) were evaluated, 32 in the transplant group and 35 in the chronic RBC transfusions group. No deaths or strokes were observed in either group during the study. There was no HSCT rejection in the transplant group and no cases of graft-versus-host disease (GVHD). The authors reported significantly lower TCD velocities one year after follow-up in the transplant group compared to the chronic pRBC transfusions group (difference of −40.8 cm/s [95%CI, −62.9 to −18.6]; p < 0.001). Additionally, TCD normalization was higher in the transplant group compared to the transfusions group (80% versus 48% in 1 year; p = 0.045).39(A)
Given the results obtained so far, it is possible to observe the curative potential of HSCT in patients with SCA and its role in preventing stroke. As highlighted above, these data are confirmed to primary stroke prevention.
Recommendation
Patients with sickle cell anemia and abnormal conventional transcranial doppler (≥200 cm/s) should undergo HLA testing to search for family donors (parents and siblings). If there is a healthy matched related donor, HSCT is indicated. There is no contraindication for sickle cell trait donors.
The authors declare no conflicts of interest.
- 1
Clinical question, structured question (PICO), and search strategies
The question structures were done using the PICO system and considering the selected questions.
The articles were identified in the Medline (via PubMed) and manual search (including reference of references, reviews, and guidelines).
Initially, the studies were selected by title, sequentially by abstract, and finally by their full text, which was submitted to critical evaluation and extraction of results related to the outcomes.
Is there evidence that transcranial doppler should be performed in all patients with sickle cell disease for primary stroke prevention?
Patient: patients with sickle cell disease between 2-16 years
Intervention: transcranial doppler
Comparison: —
Outcome: primary stroke prevention
Descriptors: (Anemia, sickle cell) AND (Stroke) AND (Transcranial doppler)
Total articles: 383
1st selection: 78
2nd selection: 12
Is there evidence that chronic packed red blood cell transfusions can be used as a treatment to prevent primary stroke in patients with sickle cell disease?
Patient: patients with sickle cell disease between 2-16 years
Intervention: packed red blood cell transfusions
Comparison: clinical observation
Outcome: primary stroke prevention
Descriptors: (Anemia, sickle cell) AND (Blood transfusion) AND (Stroke)
Total articles: 453
1st selection: 51
2nd selection: 5
Is there evidence that hydroxyurea can be used as a treatment to prevent primary stroke in patients with sickle cell disease?
Patient: patients with sickle cell disease between 2-16 years
Intervention: treatment with hydroxyurea
Comparison: observation; blood transfusions
Outcome: primary stroke prevention
Descriptors: (Anemia, Sickle Cell) AND (Hydroxyurea) AND (Stroke)
Total articles: 243
1st selection: 34
2nd selection: 15
Is there evidence that hematologic stem cell transplantation can be used as treatment to prevent primary stroke in patients with sickle cell disease?
Patient: patients with sickle cell disease, TCD ≥ 200 cm/s, and matched related donor.
Intervention: hematologic stem cell transplantation
Comparison: clinical observation
Outcome: primary stroke prevention
Descriptors: (Anemia, sickle cell) AND (Stroke) AND (Transplant)
Total articles: 174
1st selection: 20
2nd selection: 4
- 2.
Initial eligibility criteria for studies
- •
Components of PICO
- •
No time limit
- •
Portuguese, English, French, or Spanish
- •
Full-text availability
- •
- 3.
Selection of articles
Studies were selected independently and blinded after evaluating titles and abstracts of articles obtained using the search strategy, strictly obeying the inclusion and exclusion criteria to separate studies of potential relevance. When the title and abstract were not informative, the article was read in full.
The main reasons for exclusion were that they did not fulfill the PICO criteria and intermediate outcomes. Narrative reviews, case reports, case series, and preliminary results were excluded. As the question is about treatment, the option was the type of study: comparative observational studies (cohort and/or before and after) and comparative experimental studies (clinical trials).
- 4.
Critical evaluation and strength of evidence
The strength of evidence from observational and experimental studies was defined considering the study design and the corresponding risk of bias, the analysis results (magnitude and precision), the relevance, and the applicability (Oxford/GRADE).1,40
The level of Scientific Evidence was classified by type of study according to the Oxford criteria as:1
A: Major experimental and observational studies
B: Minor experimental and observational studies
C: Case reports (non-controlled studies)
D: Opinion without critical evaluation based on consensus, physiological studies, or animal models
If the article selected in the search was defined as a randomized controlled clinical trial (RCT), it was submitted to an appropriate critical assessment checklist (Table 2). The critical evaluation of the RCT allows classification according to the JADAD score, considering a JADAD score <3 as inconsistent and articles with a score ≥ of 3as consistent.41
Critical outline of randomized controlled trials (checklist).
When the selected article was defined as a comparative study (observational cohorts or non-randomized clinical trials), it was submitted to an appropriate Critical Evaluation Checklist (Table 3) with the classification of the study being according to the Newcastle-Ottawa scale, considering cohort studies consistent with score ≥ 6 and inconsistent <6.42
Scheme of critical evaluation of cohort studies.



