



So far, more than 800 mutations involving the beta-globin gene have been characterized worldwide. Most of these variants are also carried by patients with Turkish origin, since Turkey is one of the hotspot regions for mutations of the globin genes.1
In the present study, we report a case with an unusual high-performance liquid chromatography (HPLC) pattern who turned out to be a carrier of the ¿78(EF2) Leu>Val; HBB: c.235 C>G mutation. This mutation is previously unreported in Turkish individuals.
Case reportThe patient attended our laboratory for hematological exams due to persistent anemia and hemoglobinopathy HPLC analysis was ordered as hemoglobinopathies are common in patients of Turkish descent.
The proband was a 70-year-old female from the eastern Black Sea region.
The complete blood count of the proband revealed a red blood cell (RBC) count of 3.36×103/L, hemoglobin (Hb) level of 8.7g/dL, mean corpuscular volume (MCV) of 81.1fL and mean corpuscular Hb (MCH) of 25.8pg (Table 1).
The hematological and molecular data of the patient.
| Parameter | Proband | Reference range |
|---|---|---|
| Age (years) – gender | 70, F | – |
| Hb (g/dL) | 8.7 | 13.0–17.0 |
| RBC (×103/L) | 3.36 | 4.0–5.2 |
| MCV (fL) | 81.13 | 80–92 |
| MCH (pg) | 25.84 | 27–31 |
| RDW-CV (%) | 11.88 | 11.5–14.5 |
| Hb A (%): retention time/min | 35.01: 2.618 | 94–99 |
| Hb A2 (%): retention time/min | 3.99: 3.141 | 0–3.90 |
| Unknown peak (%): retention time/min | 57.31: 2.701 | – |
| Serum LDH level (U/L) | 257 | 90–241 |
| Serum iron level (¿g/dL) | 32 | 40–170 |
| ¿-Globin genotype | ¿c.235C>G/¿A | ¿A/¿A |
Hb: Hemoglobin; RBC: red blood cell count; MCV: mean corpuscular volume; MCH: mean corpuscular hemoglobin; RDW: red blood cell distribution width; LDH: lactate dehydrogenase.
Reference ranges used are based on the equipment manufacturers’ established values for the gender and age group of the proband.
HPLC analysis was performed using an in-house method employing an ultra-speed HPLC system with a 415nm UV/VIS-Detector (Shimadzu Corp., Japan). The method to obtain a Hb chromatogram was modified from Ou et al.2 The efficiency of the system is controlled prior to every analysis using a ready-to-use test solution containing the Hb variants Hb A1c, Hb F, Hb A, Hb E, Hb A2, Hb D, Hb S and Hb C, and control hemolysates at two levels of concentration, each containing Hb F, Hb A, Hb A2 and Hb S (RECIPE Chemicals+Instruments GmbH, Germany).
In the HPLC analysis, the Hb A level was 38.8%, with a slightly increased Hb A2 level (3.99%). No Hb F peak was seen in the first and following analyses. Besides this, an unknown 57.3% peak was identified in the Hb E window adjacent to the Hb A peak, indicating a possible heterozygous hemoglobin mutation. The sample was rerun and exhibited an identical pattern (Figure 1).
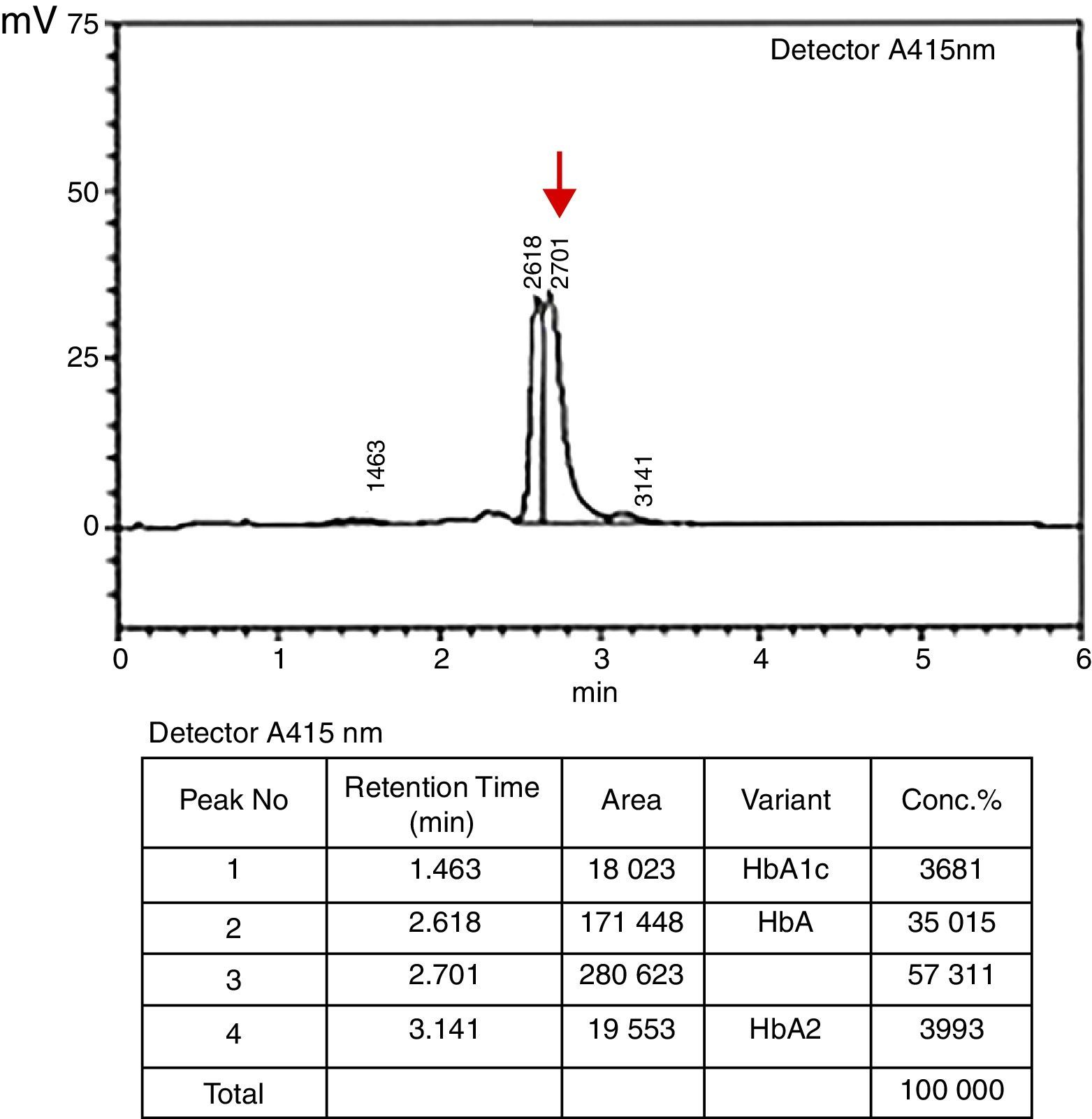
Pattern of the patient with ¿78(EF2) Leu>Val; HBB: c.235 C>G mutation using an in-house high-performance liquid chromatography (HPLC) system. In our method, a reference HPLC pattern comprises Hb A1c, Hb F, Hb A and Hb A2 chains respectively with their order of appearance on the chromatogram. However, an abnormal variant peak (indicated with an arrow) was observed in the proband on the 2.7th minute of the migration through the column with a concentration of 57.3%. Additionally, Hb A2 level (retention time: 3.141min) of the proband was slightly increased (3.99% compared to the reference range of 0–3.90%).
In order to have a detailed analysis of the suspected mutation, the beta globin gene was amplified by polymerase chain reaction (PCR) and the PCR product was analyzed by direct nucleotide sequencing using a Beckman Coulter CEQ 2000XL DNA sequencer.
DNA sequencing of the entire beta globin gene and the promoter region revealed the proband was heterozygous for the ¿78(EF2) Leu>Val; HBB: c.235 C>G mutation, named Hb Ullevaal as described by Grimholt et al.3 (Figure 2).
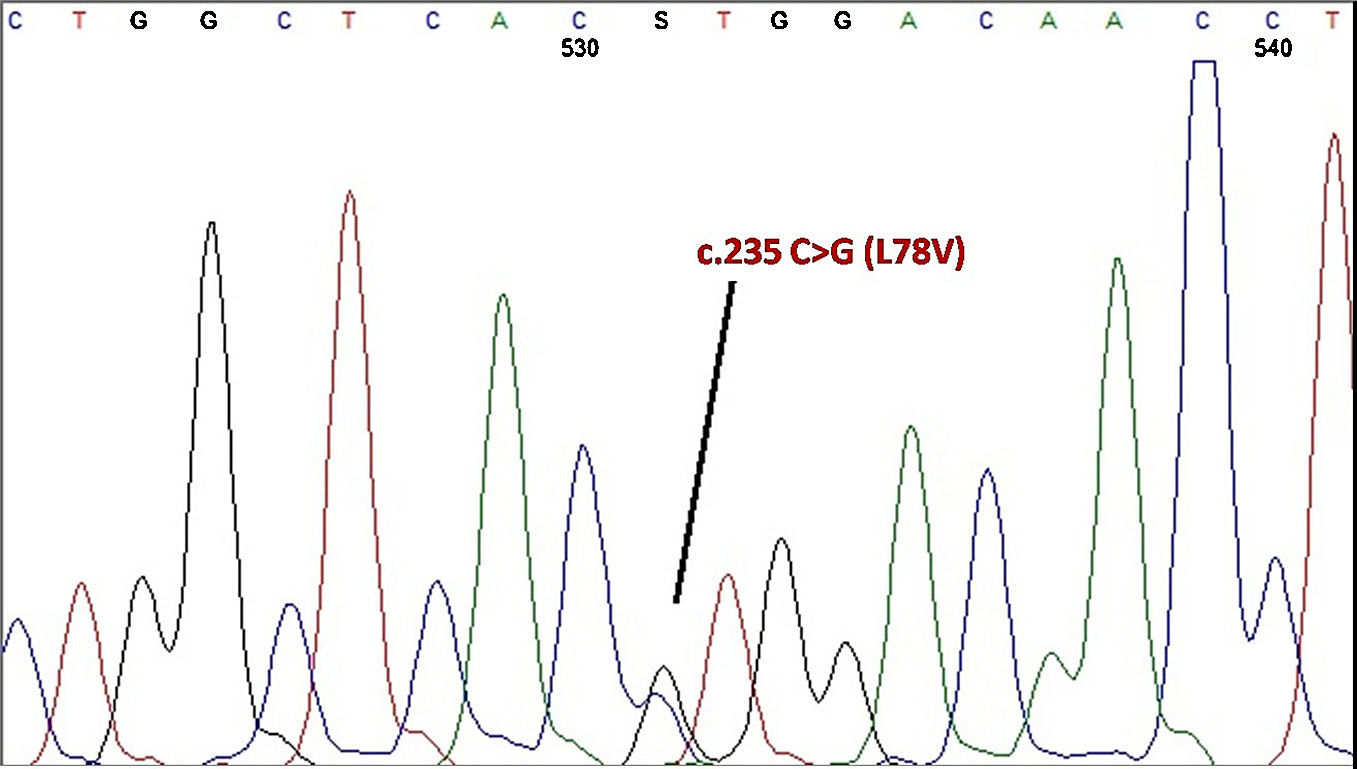
This variant has previously been reported in only one person, a 61-year-old female Norway resident from Bosnia, a community with cultural and economical aspects similar to Turkey since the Ottoman Empire, suggesting a historical path for the mutation. Although no abnormal pattern was observed by HPLC analysis and the mutation was detected incidentally when the patient was referred for the measurement of Hb A1c by cation exchange HPLC, the variant Hb peak resided between Hb A and Hb A2 on the HPLC chromatogram in the first report defining this substitution. Additionally, moderate anemia with decreased RBC, Hb, MCV, MCH and iron levels and a slightly elevated LDH concentration was present in the proband.
Unfortunately, we were not able to analyze other family members genetically. However, sequence analysis involving the alpha globin gene and beta-globin gene promoter region resulted in no variations.
As Leucine and Valine are both basic amino acids and this substitution does not change the net charge of the beta-globin molecule, a computerized analysis using the Garnier Osguthorpe Robson (GOR) method for protein structure prediction software revealed an altered beta helix structure. The loop angle corresponding to the substituted amino acid region was narrower assuming a conformational change in the protein.
The beta globin gene cluster haplotypes of this mutation are linked to haplotype 1 (+−−−−++) in our case.4
In conclusion, as the first report of the described mutation suggests there should be no anemia in the proband, the anemia seen in this case might be a result of iron deficiency or other conditions. This very first report of the ¿78(EF2) Leu>Val; HBB: c.235 C>G mutation in a Turkish individual highlights the great diversity of the beta globin gene present in the Turkish population and contributes knowledge of the mutational relationship in a ‘bench top archeological’ manner between different communities due to historical connections.
Conflicts of interestThe authors declare no conflicts of interest.



