




Adoptive transfer of T cells expressing a CD19-specific chimeric antigen receptor (CAR) has shown impressive response rates for the treatment of CD19 + B-cell malignancies in numerous clinical trials. The CAR molecule, which recognizes cell-surface tumor-associated antigen independently of human leukocyte antigen (HLA), is composed by one or more signaling molecules to activate genetically modified T cells for killing, proliferation, and cytokine production.
ObjectivesIn order to make this treatment available for a larger number of patients, we developed a simple and efficient platform to generate and expand CAR-T cells.
MethodsOur approach is based on a lentiviral vector composed by a second-generation CAR that signals through a 41BB and CD3-ζ endodomain.
ConclusionsIn this work, we show a high-level production of the lentiviral vector, which was successfully used to generate CAR-T cells. The CAR-T cells produced were highly cytotoxic and specific against CD19+ cells in vitro and in vivo, being able to fully control disease progression in a xenograft B-cell lymphoma mouse model. Our work demonstrates the feasibility of producing CAR-T cells in an academic context and can serve as a paradigm for similar institutions. Nevertheless, the results presented may contribute favoring the translation of the research to the clinical practice.
Progress in genetic engineering technology has simplified the generation of antitumor T lymphocytes. By artificially introducing an antigen-specific T cell receptor (TCR) into cytotoxic T lymphocytes, the affinity to tumor cells can be increased.1 However, this approach still depends on the presentation of the target antigen by the major histocompatibility complex (MHC) of the tumor cell, a limitation that can be overcome by the introduction of a synthetic recognition framework called the chimeric antigen receptor (CAR).2 Cell therapy using CAR-T lymphocytes is an emerging immunotherapeutic approach to treat a variety of neoplastic diseases, including lymphomas and leukemias. These CAR-T cells recognize molecules present on the surface of tumor cells, independent of the MHC system, making the antitumor response more effective3,4 This independence of MHC allows CAR-T cells to be used to treat any patient whose tumor expresses the target antigen.
Several gene transfer platforms have been developed and are available to introduce the CAR transgene into primary T lymphocytes. Most of the current studies use retroviral vectors, such as lentiviral and γ-retroviral vectors.5 Lentiviral vectors have become particularly attractive for clinical applications due to their ability to efficiently transduce most cell types, including non-proliferating cells such as Naive T cells. The major advantage of utilizing a lentiviral vector-based approach is that fewer patient-derived T cells are required for successful transduction and expansion to achieve the target dose for clinically relevant infusion. These characteristics make lentiviral vectors an attractive tool for the engineering of CAR-T cells capable of generating robust clinical responses even in patients with advanced B cell malignancies.6
Therapies with anti-CD19 CAR-T lymphocytes have shown excellent results in patients with B lymphocyte neoplasias, inducing remission in children and young adults with lymphoid leukemia.7–11 Clinical trials in chronic lymphocytic leukemia (CLL) show that anti-CD19 CAR-T lymphocytes containing the 4-1BB co-stimulation domain successfully proliferate in vivo, eliminating the disease and exhibiting functional activity for more than 3 years.12 Studies of 3 different groups report remission rates of 70%–90% in acute lymphocytic leukemia (ALL) patients treated with anti-CD19 CAR-T lymphocytes.8,10,11
Despite the impressive results, this therapeutic approach has limited availability due to complexity and cost. There are only two approved commercial products, Kymriah of Novartis and Yescarta of KITE Pharma, used to treat B-cell ALL and diffuse large B-cell lymphoma (DLBCL). However, the treatment cost associated with these products is restrictively high in most cases (US$475,000 and US$373,000, respectively), thus limiting widespread patient access. For this reason, we developed a simple and efficient platform for the production of lentiviral vectors and CAR-T cells. Here we describe a high-level production of a lentiviral vector encoding CD19-CAR expression, consisted of a transmembrane domain CD19 scFv (MCF63) and intracellular 4-1BB-CD3ζ signalling domains. The generated CAR-T cells exhibited expansion capability and antitumoral in vitro and in vivo efficacy.
MaterialEthical approvalThis research was approved by the Ethical Review Board of the Clinical Hospital, Ribeirão Preto Medical School, University of São Paulo (Protocols 1.996.240 and 2.053.927) and by the National Commission for Research Ethics (CONEP, Protocols 2.183.633 and 2.183.143). All subjects signed informed written consent in compliance with the Resolution 466/2012 of the Brazilian National Health Council (CNS). The use of animals in this research has been approved by the Local Animal Ethical Committee at the Ribeirão Preto Medical School (Protocol 124/2017).
Lentiviral vector productionLentiviral vector production was generated by the transient cotransfection of HEK 293 T cells with a four-plasmid system: pCAR19, gene expression cassette for anti-CD19 antigen chimeric receptor and 4-1BB costimulatory domain; LentiArt™ pHelp1, capsid cassette containing the gag, pol and RRE viral genes; LentiArt™ pHelp2 VSV-G viral envelope cassette; and, LentiArt™ pHelp3, capsid cassette containing the viral gene Rev (Creative Biolabs). The HEK293 T/17 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Gibco), supplemented with 10% fetal bovine serum (FBS, Hyclone). A T175 cm2 monolayer culture with 60–80% confluency was transfected with 60 μg of plasmid DNA in a 3:1:1:1 or 4:2.6:1.4:1 ratio (transgene:gag-pol:VSV-G:rev), with 180 μg Polyethyleneimine (PEI, Alfa Aesar) or Lipofectamine® 2000 (Life Technologies), according to manufacturer instructions. Viral supernatant was collected by using 3 different approaches: 24 h post-transfection; 48 h post-transfection; or 24 h post-transfection, followed by the addition of fresh medium and another collection 48 h post-transfection. The addition of sodium butyrate at the time of transfection at a final concentration of 5 mM.
The vector particles in the supernatant were filtered through a 0.45 μm filter and three concentration methods were evaluated: i) ultracentrifugation at 19,200 rpm for 1 h 40 min at 4 °C in Optima™ XL-100 K ultracentrifuge (Beckman Coulter, rotor SW28, equivalent to approximately 67,000g), ii) tangential flow filtration with a 100 kDa cartridges. The cartridges were coupled to QuixStand apparatus (GE Healthcare), according to the manufacturer’s instructions followed by ultrafiltration through a 100 kDa molecular weight cut off Vivaspin concentration tube (GE Healthcare) and iii) tangential flow filtration with a 100 kDa cartridges followed by ultracentrifugation. The viral pellet was resuspended in the Roswell Park Memorial Institute (RPMI-1640, Gibco) medium, supplemented with 10% AB human serum and 0.0001% Pluronic® F-68 (Gibco). AB human serum was prepared as described previously 13 viral titration was performed by flow cytometry after transduction of the Jurkat cell line. The detection of CAR on the cell surface was assessed by Alexa Fluor® 647-conjugated anti-F(ab')2 antibody (Jackson Immunoresearch).
Generation and expansion of CAR-modified human T lymphocytesTwenty million CD3+ T cells, isolated and selected from the Peripheral Blood Mononuclear Cells (PBMC) of healthy donors, were stimulated with anti-CD3/CD28 mAb immobilized on magnetic beads (Dynabeads™ Human T-Activator CD3/CD28 for T Cell Expansion and Activation, Gibco) at a 1:1 ratio (bead:cell). The next day, cells were transduced with the lentivirus (MOI 5) by overnight incubation in the presence of polybrene (Santa Cruz Biotechnology, sc-134220) at 8 mg/mL. Cells were expanded in the complete medium (RPMI supplemented with 10% AB human serum and 100 U/mL interleukin 2, or IL-2) in a 6-well plate and incubated at 37 °C in a humidified incubator, 5% CO2. Cell proliferation was maintained by the addition of fresh media on days 4, 7 and 10. On day 7, the activation beads were removed. Cell numbers and viability were determined at each time point by trypan blue exclusion. Throughout expansion, cells were transferred to appropriate culture plates and flasks, as needed, to maintain a cell concentration of 1 × 106 cells/mL.
Phenotypic characterization of expanded CD19-CAR-T cellsTo determine the phenotypic characteristic of CD19-CAR-T cells, the expression of the surface markers was analyzed by the flow cytometry. The cells were washed three times with Phosphate-Buffered Saline (PBS) containing 0.5% Goat Gamma Globulin and centrifuged for 3 min at 1800 rpm. Subsequently, cells were washed with PBS, centrifuged for 3 min at 1800 rpm, resuspended in 100 µL of PBS and incubated with 1 µL of Alexa Fluor 647 ChromPure Goat IgG (1/100) and 1 µL Alexa Fluor 647 AffiniPure F(ab') Fragment Goat Anti-Mouse IgG (1/100) on ice for 45 min in the dark. Finally, cells were washed with 1 mL of PBS, centrifuged for 3 min at 1800 rpm and then resuspended in PBS and analyzed by flow cytometry (FACSAria, Becton-Dickinson, San Diego, CA, USA).
In vitro cytotoxicityCytotoxic activity of generated CD19-CAR-T cells was evaluated by flow cytometry analysis and by LDH (lactate dehydrogenase) assay. For FACS analysis, CD19+ and CD19- target cells (2 × 106) were pre-stained with the green fluorescent membrane dye PKH67-GL (Sigma–Aldrich) and effector cells (CAR T- cells and non-transduced T cells) were added to 5 × 105 target cells to yield effector to target (E:T) ratios of 10:1. The culture was maintained in RPMI supplemented with 10% AB serum and 100 IU/mL IL-2 at 37 °C and 5% CO2. The percentage of target cells was assessed by flow cytometry on day zero and after 24 h of incubation. The cell mixture was centrifuged at 260g and stained with propidium iodide (PI, 5 μg/mL). Dead target cells were identified by simultaneous PKH67-GL and PI-positive. Target cells incubated without effector cells were used to assess spontaneous cell death. Cytotoxicity was also assessed using the Pierce LDH Cytotoxicity Assay kit (Thermo Fisher). The E:T ratios surveyed were 1:1 and 5:1. Target and effector cells were maintained in a complete RPMI medium at a final concentration of 1 × 105 to 3 × 105 cells/mL. The LDH release was measured in the supernatant, according to the manufacturer’s instructions. The maximum LDH release was measured in target cells incubated with the provided 10X lysis buffer. Target cell cytotoxicity was calculated using the following formula: %Cytotoxicity = 100*[(CAR+:Sup-B15 – CAR + alone – target alone)/(Maximum target lysis – target alone)].
B-cell lymphoma xenograft murine modelEight-week-old NSG-SGM3 (NOD.Cg-PrkdcscidIl2rgtm1Wjl Tg[CMV-IL3,CSF2,KITLG]1Eav/MloySz; Jackson Laboratory) female mice were injected subcutaneously in the right flank with 5 × 106 Raji luciferase positive (Raji-Luc) cells in 0.1 mL of saline solution. Eleven days later, mice bearing subcutaneous tumors were infused (single IV) with 4 × 106 CD19-CAR-T cells or PBS as a control. The animals were then monitored three times a week for weight and tumor growth (measuring 3 perpendicular diameters). Tumor progression was also confirmed by BLI (bioluminescence imaging) using a Xenogen IVIS LUMINA III in vivo Imaging System (PerkinElmer) for up to 40 days. The animals were killed when tumor volume reached a dimension of 1000 mm3, before they exhibited clinical signs and symptoms to avoid unnecessary pain and discomfort, according to standard ethical animal guidelines.
Quality control proceduresMycoplasma contamination of cultured cells was determined by the MycoAlert® (Lonza), according to the manufacturer's instructions, using a luminometer (Victor X3, Perkin Elmer). Briefly, ATP (adenosine triphosphate) levels in each sample were measured before (Reading A) and after (Reading B) the addition of the Mycoalert® substrate. This ratio (B/A) was considered an indicator for the presence of mycoplasma contamination. The presence of contamination was proved if the Reading B/Reading A ratio was greater than 1. The procedure was performed according to the manufacturer's instructions and was measured using a luminometer (Victor X3, Perkin Elmer).
Endotoxin levels were measured using the Charles River Endosafe®-PTS™ system, according to the manufacturer's instructions. All samples tested with the Endosafe system used 0.05–5.0 endotoxin unit/mL (EU/mL) sensitivity cartridges provided by Charles River.
The BacT/ALERT®3D microbial detection system (BioMérieux, Paris, France) was used to detect aerobic and anaerobic bacterial contamination in the cell cultures.
ResultsHigh-yield production of lentiviral vectorLentiviral vectors are the most commonly used method to generate CAR-T cells in clinical trials. In this study, we first developed an efficient platform for lentiviral production. The CD19-directed CAR expression cassette was composed of an anti-CD19 scFv fused to a CD8α hinge and transmembrane region and the intracellular signaling domains of human 4-1BB and CD3ζ motif in tandem.
To optimize the HEK293 T cell transfection, we evaluated four factors that could potentially affect transfection efficiency and lentivirus output: (1) the ratio of four plasmids; (2) the time of harvesting the viral particles; (3) the type of transfection reagent; and, (4) the concentration method.
In the four-plasmid lentivirus production system, the ratio of plasmids is crucial to the yield of lentivirus. Diverse combinations have already been described.14–16 To optimize this variable, we examined the lentivirus output with two different combinations of pCAR:gag-pol:VSVg:rev plasmids: a) 3:1:1:1 and b) 4:2.6:1.4:1. As shown in Figure 1A, the largest proportion (4: 2.6:1.4:1) did not result in a significant increase in the viral titer obtained. So, we decided to use the 3:1:1:1 plasmids ratio for the next experiments. Regarding the best harvesting time, we obtained the highest lentivirus production when 24 -h and 48 -h collections were combined; the first harvest being made 24 h after transfection, followed by the addition of fresh culture medium and a second collection after 48 h of transfection (Figure 1B).
 Viral titer (TU/mL) using different plasmid ratios (transgene:gag-pol:VSV-G:rev). Data presented as mean ± SD, n = 2. Non-significant difference (Mann-Whitney U-test). (B) Effect of viral supernatant harvesting times on Viral titer (TU/mL). Single harvests at 24 and 48 h post-transfection (hpt), and two harvests at 24 and 48 hpt. Data presented as mean ± SD, n = 3. **P < 0.002 (ANOVA followed by Holm-Sidak). (C) Effect of transfection agent on Viral titer (TU/mL). Polyethyleneimine (PEI) and Lipofectamine® 2000 (Lipofectamine) were employed in the absence or presence of NaBu (5 mM). Data presented as mean ± SD, number of replicates indicated by number of points, *P < 0.05; **P < 0.002; ***P < 0.0002; ****P < 0.0001 (ANOVA followed by Holm-Sidak). (D) Effect of concentration methods on lentiviral recovery. Ultracentrifugation (UC), tangential flow filtration (QuixStand - QS), tangential flow filtration, followed by ultracentrifugation (QS + UC), ultrafiltration by membrane centrifugation (VivaSpin - vs) were employed. Data presented as mean ± standard deviation, number of replicates indicated by the number of points, **P < 0.002; ****P < 0.0001 (ANOVA followed by Holm-Sidak).")
Establishment of improved conditions for lentiviral production. Effect of plasmid ratio on viral production: (A) Viral titer (TU/mL) using different plasmid ratios (transgene:gag-pol:VSV-G:rev). Data presented as mean ± SD, n = 2. Non-significant difference (Mann-Whitney U-test). (B) Effect of viral supernatant harvesting times on Viral titer (TU/mL). Single harvests at 24 and 48 h post-transfection (hpt), and two harvests at 24 and 48 hpt. Data presented as mean ± SD, n = 3. **P < 0.002 (ANOVA followed by Holm-Sidak). (C) Effect of transfection agent on Viral titer (TU/mL). Polyethyleneimine (PEI) and Lipofectamine® 2000 (Lipofectamine) were employed in the absence or presence of NaBu (5 mM). Data presented as mean ± SD, number of replicates indicated by number of points, *P < 0.05; **P < 0.002; ***P < 0.0002; ****P < 0.0001 (ANOVA followed by Holm-Sidak). (D) Effect of concentration methods on lentiviral recovery. Ultracentrifugation (UC), tangential flow filtration (QuixStand - QS), tangential flow filtration, followed by ultracentrifugation (QS + UC), ultrafiltration by membrane centrifugation (VivaSpin - vs) were employed. Data presented as mean ± standard deviation, number of replicates indicated by the number of points, **P < 0.002; ****P < 0.0001 (ANOVA followed by Holm-Sidak).
As demonstrated in Figure 1C, the lentiviral titers produced were significantly different between the PEI- and Lipofectamine-mediated transfection methods. Lipofectamine enabled a production 3 times higher than PEI. In both cases, the addition of sodium butyrate during transfection was beneficial (p < 0.05) in the production of high-titer lentivirus.
Three methods for lentiviral concentration were tested in this study: ultracentrifugation, tangential filtration and ultrafiltration (Figure 1D). The recovery of lentiviral particles did not exceed 54% and 61%, when using ultracentrifugation and tangential filtration, respectively. Moreover, post-filtration volumes were still large (ranging from 50 to 134 mL) and low in titers (approx. 105 TU/mL, data not shown). As an attempt to decrease the volume and increase the viral titer after the tangential filtration, ultracentrifugation was performed. Combining the two methodologies, our recovery rate did not reach 26% of viable particles. To decrease the loss of concentrated viral particles, we then evaluated the ultrafiltration concentration method using a 100 kDa molecular weight cutoff (MWCO). The average yield of the ultrafiltration concentration was 81.8%, significantly higher than the other concentration methods tested.
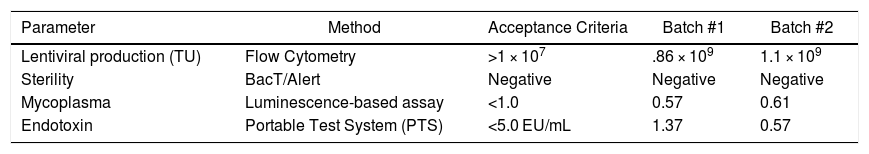
Using the best conditions defined above, two batches were produced and submitted to quality control (Table 1). Each lentiviral batch was concentrated and cryopreserved at -80 °C. The viral titer of frozen-concentrated virus ranged from 1.1 to 2 × 109 TU/mL. The protocol established enabled the production of a clinical-grade product (tested for bacteria, fungus and mycoplasma). Moreover, endotoxin levels were below the values recommended by regulatory agencies (<5.0 EU/mL).
Quality control of the lentiviral vector produced 24 and 48 h after transfection using a plasmid ratio of 3:1:1:1 (transgene:gag-pol:VSV-G:rev) and lipofectamine. Particle concentration was performed by membrane ultrafiltration (VivaSpin™ 100 kDa, GE Healthcare).
| Parameter | Method | Acceptance Criteria | Batch #1 | Batch #2 |
|---|---|---|---|---|
| Lentiviral production (TU) | Flow Cytometry | >1 × 107 | .86 × 109 | 1.1 × 109 |
| Sterility | BacT/Alert | Negative | Negative | Negative |
| Mycoplasma | Luminescence-based assay | <1.0 | 0.57 | 0.61 |
| Endotoxin | Portable Test System (PTS) | <5.0 EU/mL | 1.37 | 0.57 |
Enriched CD3 + T cells derived from healthy donors were activated with CD3/CD28 beads (ratio 1:1) and transduced with the lentiviral vector (MOI 5) to express the CD19-specific chimeric antigen receptor according to the protocol presented in Figure 2A. T cells were expanded in vitro in static flasks for 10 days in the presence of interleukin (IL-2). This protocol enabled the production of 5.1 ± 0.14 × 108 cells at day 10 (Figure 2B), representing a 22.6 ± 4.79-fold increase (Figure 2C). At the beginning of the expansion (Day 4), transduction efficiency of approximately 20% was obtained, increasing during the time in culture (40% of the cells were CAR+).
In vitro specificity and cytolytic potency of CD19-CAR-T cells against target CD19 + B cells Schematic representation of CD19-CAR-T cells production protocol. (B) Cell growth profile of CD19-CAR-T cells (n = 2) expanded with 100U/mL IL-2. (C) Fold increase in CD19-CAR-T cell number. (D) Percentage of CD19-CAR-T cells expression during the time of culture.")
We evaluated the specific cytolytic function of CD19-CAR-T cells produced by two methodologies: flow cytometry and colorimetric assay (LDH). To assess cytotoxicity, we incubated CD19-CAR-T cells with the human CD19+ cell line Sup-B15 and CD19- K562 cell line. The CD19-CAR, or non-transduced T cells, were co-cultured with Sup-B15 or K562 target cells. The ratio 10:1 (effector:target) was used and the target cell percentages present at the beginning and 24 h after co-culture were evaluated by flow cytometry (Figure 3A).
 Illustrative dot plots of co-cultivation analysis (0 and 24 h) by flow cytometry. Effector cells: non-transduced T cells (T) and CAR-T cells (CAR). Target cells (PKH67+): Sup-B15 (CD19+) and K562 (CD19-). (B) Percentage of target cells (PKH67+) at the beginning and after 24 h of co-cultivation. Data presented as mean ± SD, n = 4. *P < 0.05, ****P < 0.0001 (ANOVA two factors followed by Holm-Sidak). (C) Cytotoxicity percentage calculated by the absorbance related to LDH released in the co-cultures in the 1:1 and 5:1 (effector: targets) proportions. Effector cells: non-transduced T lymphocytes (T) and CAR-T lymphocytes (CAR). Sup-B15 (CD19 +) and K562 (CD19-) target cells. Data presented as mean ± SD, n = 3. ****P < 0.0001 (ANOVA two factors followed by Holm–Sidak).")
(A) Illustrative dot plots of co-cultivation analysis (0 and 24 h) by flow cytometry. Effector cells: non-transduced T cells (T) and CAR-T cells (CAR). Target cells (PKH67+): Sup-B15 (CD19+) and K562 (CD19-). (B) Percentage of target cells (PKH67+) at the beginning and after 24 h of co-cultivation. Data presented as mean ± SD, n = 4. *P < 0.05, ****P < 0.0001 (ANOVA two factors followed by Holm-Sidak). (C) Cytotoxicity percentage calculated by the absorbance related to LDH released in the co-cultures in the 1:1 and 5:1 (effector: targets) proportions. Effector cells: non-transduced T lymphocytes (T) and CAR-T lymphocytes (CAR). Sup-B15 (CD19 +) and K562 (CD19-) target cells. Data presented as mean ± SD, n = 3. ****P < 0.0001 (ANOVA two factors followed by Holm–Sidak).
After 24 h of co-incubation, CD19-CAR-T cells eliminated 87% of Sup-B15 CD19+ cells (15.24% to 1.91% of Sup-B15 CD19+ cells). In the co-culture of non-transduced lymphocytes with Sup-B15 cells, no difference was observed. Similarly, there was no death of CD19- cells (K562) after co-culture with CD19-CAR-T cells, demonstrating the specificity of the CAR to the CD19 antigen.
The cytotoxicity of CD19-CAR-T cells was next evaluated by the LDH release assay. As shown in Figure 3C, CD19-CAR-T cells were able to lyse the Sup-B15 (CD19+), but no toxicity was observed against K562 (CD19-) cells. Taken together, these results demonstrate that the CD19-CAR-T cells produced have effective antigen-specific cytolytic activity.
Antitumoral efficacy of CD19-CAR-T cells in Raji B-cell lymphoma xenograft modelIn order to evaluate and validate the therapeutic efficacy of CD19-CAR-T cells in vivo, we established a murine model of B-cell lymphoma. To reach this purpose, Burkitt lymphoma Raji cells (ATCC® CCL-86™) were transduced with a lentiviral vector containing the luciferase gene (RajiLuc+) for real-time evaluation of disease progression.
The NSG-SGM3 mice were injected subcutaneously with 5 × 106 tumor cells (day 0) and the luciferase activity was evaluated in vivo on days 4, 11, 14, 18, 23, 28, 32, 36, 44, 47 and 50 (Figure 4A, B and C). Tumor growth was also evaluated by pachymeter. The CD19-CAR-T cells (4 × 106 cells) were infused at day 11. Tumor size evaluation was performed until day 29 (Figure 4D), upon which the animals of the control group were euthanized due to the exacerbated tumor volume. Mice that received CD19-CAR-T cells presented a greater control of tumor growth. In summary, the CD19-CAR-T cells produced have significant in vivo cytolytic/cytotoxic activity (the anti-tumoral effect), corroborating with the in vitro results.
 Upper panel shows a timeline of experimental design. On day 0, NSG mice were injected intravenously with 5 × 106 Raji-Luc + cells. On day 11, the mice received 1 × 106 CD19-CAR-T cells or PBS. (B) Bioluminescent imaging pre-treatment (Day 5), and on day 11, intravenous injection of CD19-CAR-T cells or PBS. Disease progression was followed until day 50. (C) Tumor volume monitoring in treated and control mice.")
Evaluation of CD19-CAR-T cells infusion in a xenograft mouse model. (A) Upper panel shows a timeline of experimental design. On day 0, NSG mice were injected intravenously with 5 × 106 Raji-Luc + cells. On day 11, the mice received 1 × 106 CD19-CAR-T cells or PBS. (B) Bioluminescent imaging pre-treatment (Day 5), and on day 11, intravenous injection of CD19-CAR-T cells or PBS. Disease progression was followed until day 50. (C) Tumor volume monitoring in treated and control mice.
The transfer of specific chimeric antigen-recognition receptors into T cells offers great potential to target a specific tumor-associated antigen, such as CD19. Specifically, redirecting T cells using CAR receptors enables the efficient binding of effector T cells to tumor cells in a non–MHC-restricted fashion, thus bypassing the MHC/peptide complex loss that is a major escape mechanism found in tumors.
The use of genetically modified autologous T cells that express an anti-CD19 CAR has shown impressive response rates for the treatment of CD19-positive B-cell malignancies in several clinical trials. However, bringing this experimental treatment to the commercialization phase is a lengthy process. The urgent need to make this treatment widely available prompted us to develop a simple and efficient platform to produce anti-CD19 CAR.
The platform here developed included the lentiviral vector production step and the ex vivo transduction and expansion of genetically engineered T cells. For vector production, we chose a third-generation lentiviral vector split into a four-plasmid system because it is very efficient and is considered one of the safest viral systems to be used in clinical trials.17–19 One of the great advantages of the lentiviral system for CAR-T cells generation is that relatively small numbers of T cells isolated from a leukapheresis can be efficiently transduced and expanded by 100–1000-fold ex vivo to generate enough CAR-T cells for adoptive transfer.20
It is important to note that although the third-generation vectors have gained in safety, they have lost in terms of viral particle productivity, which greatly increases the price of cell therapy with CAR-T cells based on lentiviral vectors. Due to this, we have evaluated different conditions in order to reach a high lentiviral vector level. The best results were obtained using a plasmid ratio of 3:1:1:1 (transgene:gag-pol:VSV-G:rev), improving lipofectamine as a transfection agent. A higher particle number was obtained when the culture was harvested 24 and 48 h post-transfection, the volumes collected being mixed. Particles concentration by membrane ultrafiltration resulted in the highest recovery. The total amount of lentiviral vector produced (∼2 × 109 vectors/batch) can enable the transduction of T cells from 10 patients/viral batch. The titer obtained in this study is similar to the titers previously reported.21 Although the production is still on a small scale, the inclusion of cell suspension systems and the use of bioreactors can be considered to increase the production scale.
Currently, there are different systems to expand CAR-T cells in vitro22–24 and we have developed our cell expansion platform following the experience of our Cell-Based Therapy Center (CTC), which has produced mesenchymal cells for clinical use for more than 10 years. Despite the open characteristic of the expansion system, it has the advantage of low cost. The expansion of CAR-T cells has proved to be a robust and reproducible procedure in our hands. Our results showed that at the end of the expansion we obtained an average of 5.1 ± 0.14 × 108 cells, enough for 2.5 infusion doses, considering the ideal dose of 1 × 106 CAR+/kg and a patient weighing 80kg.25,26
We also demonstrated the potent anti-lymphoma effects of CAR-T cells in our experiments in vitro and in vivo. These findings supported that our CAR-T platform could be an alternative option for generating effective CAR-T cells for clinical use.
In conclusion, we developed a platform to produce high titers of CD19-CAR lentiviral vectors and an efficient method to generate functional CAR-T cells. Our next steps will be a more in-depth investigation of pre-clinical studies and the conduction of a clinical trial using CD19-CAR-T cells to treat B-cell malignancies.
Ethical approvalThis research has been approved by the Ethical Review Board of the Clinical Hospital, Ribeirão Preto Medical School, University of São Paulo (Protocols 1.996.240 and 2.053.927) and by National Commission for Research Ethics (CONEP, Protocols 2.183.633 and 2.183.143). All subjects signed informed written consent in compliance with the Resolution 466/2012 of the Brazilian National Health Council (CNS). The use of animals in this research has been approved by Local Animal Ethical Committee from Ribeirão Preto Medical School (Protocol 124/2017).
Conflict of interest statementThe authors declare that there is no conflict of interest regarding the publication of this article.
This study was financially supported by the FAPESP (2012/23228-4, 2016/08374-5, 2017/09491-8), CTC Center for Cell-based Therapies (FAPESP 2013/08135-2) and National Institute of Science and Technology in Stem Cell and Cell Therapy (CNPq 573754-2008-0 and FAPESP 2008/578773).



