




Diversity in Classical Hematology Research
More infoThe Glanzmann Thrombasthenia (GT) and Bernard-Soulier Syndrome (BSS) are rare hereditary disorders of platelet function. Their treatment often requires platelet transfusion, which can lead to the development of alloantibodies.
ObjectiveIn this study, we aim to develop a strategy for alloantibody detection and to describe the frequency of alloimmunization in a patient population from a single center in southeastern Brazil.
MethodsSamples from patients with GT or BSS were tested using the Platelet Immunofluorescence Test (PIFT). If a positive result was obtained, a confirmatory step using the Monoclonal Antibody Immobilization of Platelet Antigens (MAIPA) and Luminex bead-based platelet assay (PAKLx) was executed. Main results: Among 11 patients with GT, we detected the presence of alloantibodies in 5 using PIFT, with confirmation through MAIPA and PAKLx in 2 (1 anti-HLA and 1 anti-HPA), resulting in a frequency of 18.1%. Among 4 patients with BSS, PIFT was positive in 3, with confirmation by MAIPA and PAKLx in 1 (anti-HLA), showing a frequency of 25%. The two patients with anti-HLA antibodies exhibited a panel reactive antibody (PRA-HLA) testing greater than 97%.
ConclusionOur study highlights the importance of identifying platelet alloimmunization in this patient population. The proposed algorithm for platelet alloantibodies detection allows resource optimization.
The Glanzmann Thrombasthenia (GT) is a rare inherited disorder of platelet function causing quantitative or qualitative defects of the platelet membrane glycoprotein (GP) IIb-IIIa (αIIbβ3) complex.1,2 The incidence of the disease is approximately 1 per million, but this increases to 1 in 200,000 in areas of high consanguinity.3 The GT may be sub-classified using laboratory criteria into type 1 variants (< 5% GPIIb-IIIa expression), which is associated with severe bleeding and type 2 (5–20% expression of the GPIIb-IIIa) or type 3 qualitative GT variants, in which the bleeding phenotype may be less severe.1
The Bernard-Soulier Syndrome (BSS) is usually associated with an autosomal recessive inheritance that causes a defect of the platelet membrane glycoprotein (GP) Ib-IX-V complex. The incidence is similar to the GT incidence, as well as the relation to consanguinity.3 The main characteristic of the disease is macrothrombocytopenia with giant platelets often leading to misdiagnosis.
The treatment for both conditions is based on platelet transfusions, which can cause adverse events, such as allergic reactions, volume overload, transfusion-related acute lung injury and transfusion-transmitted infections. Alloimmunization against human platelet antigen (HPA) or human leucocyte antigen (HLA) epitopes is also common4 for patients requiring frequent transfusions and may be associated with the platelet refractoriness and therapeutic failure of platelet transfusion.
It is also not uncommon that GT and BSS patients develop antibodies against the portions of normal glycoproteins that are deficient in the disorders, which lead to difficulty in the transfusion support.1 The risk of antibody formation may be different, depending on the subtype of the GT, considering that patients with homozygous or compound heterozygous mutations totally lacking αIIbβ3 are more prone to develop antibodies than others with some αIIbβ3 expression.5 One small study showed the presence of anti-HPA antibodies in 81% of patients with no αIIbβ3 expression, versus 25% with less severe mutations.6 The literature is yet incomplete in reference to the risk of antibody formation in BSS, but it is probably less frequent than in the GT.7
The presence of platelet alloantibodies may cause immune platelet refractoriness, worsening the transfusion management of patients with rare inherited bleeding disorders. Recombinant activated factor VII (rFVIIa) can be used for the treatment and prevention of bleeding episodes and as prophylaxis for invasive procedures in patients with GT and alloantibodies, as well as in patients with past or present immune platelet refractoriness. Nevertheless, off-label use of rFVIIa in patients with GT who have frequent bleeding episodes is increasing, mainly to avoid alloimmunization to platelets.8
The methods to detect the platelet alloantibodies may include techniques, such as the platelet immunofluorescence test (PIFT),9 which is a screening assay for the detection of antibodies in the serum, frequently used for cross-matching the patient serum with the platelets to be transfused. It is a sensitive test, but it does not allow the identification of the specificity of the antibody. Other tests, such as the MAIPA (using ELISA technique)10 and the bead-based Luminex Platelet Assay, combine a good sensitivity and specificity,11,12 however they are more laborious and costly.
In this study, we implemented a practical and efficient strategy with the combination of methods to detect the presence of alloantibodies in the GT and BSS patients, aiming to describe their frequency in this population. We also suggest an algorithm for alloantibody detection in the GT and BSS patients.
Materials and methodsPopulationPatients with the diagnosis of Glanzmann Thrombasthenia or Bernard-Soulier Syndrome followed by the Hemostasis Outpatient Clinic at the Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (HCFMUSP) who were seen at the clinic between July 2019 and February 2020 were invited to participate in this study. Peripheral blood samples from these patients were collected to perform the antibody testing. We used three different techniques to detect alloantibodies: 1) Platelet Immunofluorescence Test (PIFT); 2) Monoclonal Antibody Immobilization of Platelet Antigens (MAIPA), and; 3) Luminex bead-based platelet assay (PAKLx), according to the algorithm in Figure 1. Data related to the transfusion history and transfusion reaction were retrieved from the blood bank (Fundação Pró-Sangue São Paulo Hemocenter, Brazil) electronic files. This study was approved by the ethical committee of the Faculdade de Medicina de São Paulo (FMUSP) and all enrolled patients signed an informed consent.
Platelet immunofluorescence test (PIFT)
Fifteen serum samples were submitted to the platelet immunofluorescence test (PIFT). The PIFT identifies antibodies directed to HLA class I and/or HPA by flow cytometry, however it does not distinguish between either of these, nor from the non-specific binding of IgG via the Fc-part to the Fc-receptor. In the present study, we tested each patient sample with four selected ABO identical and recently collected (< 72h) apheresis platelets using the PIFT, as previously described.9 The patient samples were tested immediately after collection.
Monoclonal Antibody Immobilization of Platelet Antigens (MAIPA)Samples with a positive result in the PIFT were submitted to the Monoclonal Antibody Immobilization of Platelet Antigens (MAIPA) as described by Kiefel and colleagues.10 This assay is the most sensitive and specific platelet alloantibody detection technique and is performed by the conventional enzyme-linked immunosorbent assay (ELISA) technology. A platelet suspension was sensitized with serum from the patient or donor controls, washed once and then incubated with the MoAb against the GPII-IIIa, GPIaIIa, GPIbIX and HLA class I antigen (β2-microglobulin). After washing and adding alkaline phosphatase-labeled anti-human IgG, the amount of bound antibody was determined by the conventional ELISA technology. Results were calculated as a ratio of their optical density (OD), compared to that of the mean value from control sera. The specificity of platelet alloantibodies was assessed with a panel of donors typed for the HPA-1a, -1b, -2a, -2b, -3a, -3b, 5a and -5b.
Luminex bead-based platelet Assay (PAKLx)Samples with a positive result in the PIFT were also submitted to the bead-based platelet assay PAKLx (Immucor, USA). The assay is a bead-based technique for the detection and specification of antibodies against antigens located on the HPA-1, -2, -3, -4, -5, GPIV and HLA class I (beads coated with affinity purified GPs from the HPA-typed platelet donors and the HLA class I obtained from the platelet pool) using the Luminex technology.
Samples that were positive for the anti-HLA were tested in a single antigen microsphere assay (SAB). The SAB uses the Luminex technology, which is based on fluorescent microspheres coated with the HLA class I antigens from different donors (antigen panel), to qualitatively detect the IgG antibodies (Lifecodes LSA class I Immucor, USA). The analysis was performed using the antibody reactivity panel (PRA) of the Luminex multi-analyte technology (xMAP). The results were determined by the mean fluorescence intensity (MFI) of each microsphere, compared to the microspheres of the negative and positive controls.
ResultsPatient characteristicsAmong 30 patients with GT and 9 patients with BSS followed by the Hemostasis Outpatient Clinic at the Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (HCFMUSP), between July 2019 and August 2020, 11 patients with GT and 4 patients with BSS had an appointment at the clinic and were included in the study. The mean age of the patients was 35 years (from 19 to 53) for GT; 81.8% (n = 9) were females and 18.2% (n = 2) were males. We identified 6 (54.5%) patients with type I GT and 3 with type III variant GT (27.2%). The type was not available for 2 patients with GT. For BSS, the mean age was 34.5 years (from 18 to 51) and all patients were females. The mean age at the diagnosis of the disease was 8 years old (from 2 months to 23 years old) for GT and 19.1 years (from 8 month to 47 years) for BSS. One of the patients with GT had history of parenteral consanguinity. After the review of the medical files, the most common bleeding manifestation reported by patients with GT were epistaxis (n = 9/11 patients) and gum bleeding (n = 9/11 patients), while for BSS were heavy menstrual bleedings (n = 3/4 patients). Most of the patients (81.8%, n = 9/11 for GT and 75%, n = 3/4 for BSS) presented iron deficiency.
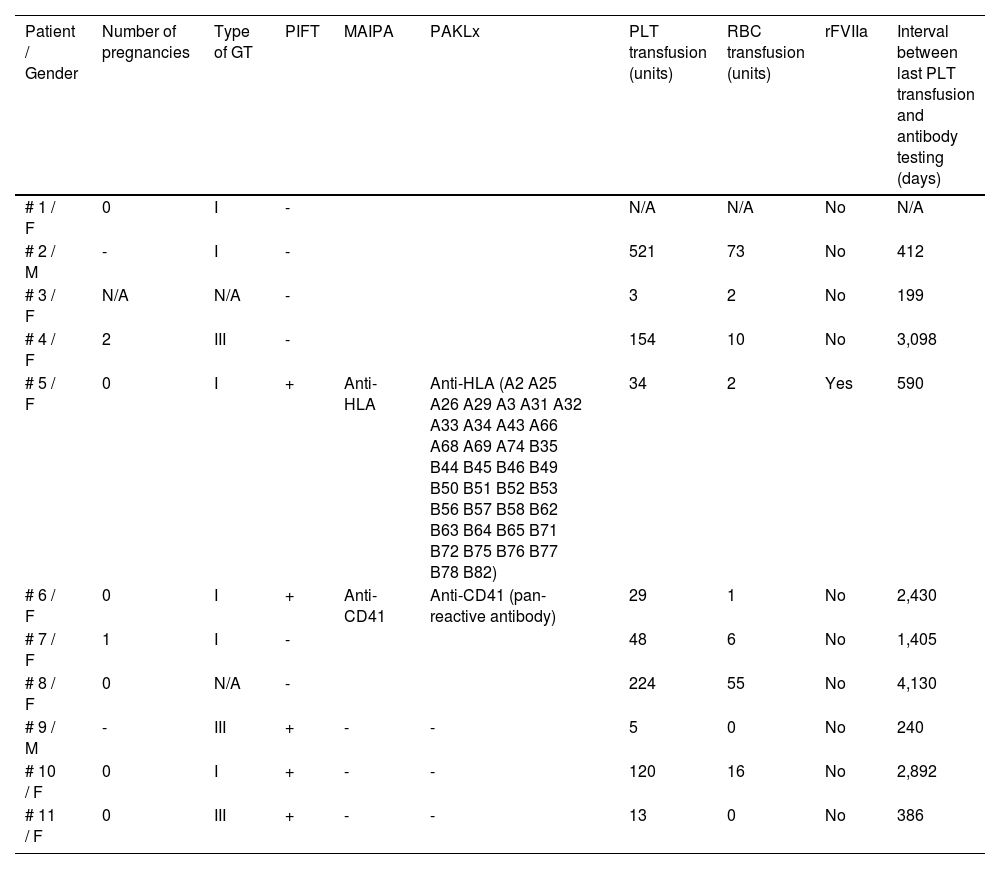
Alloimmunization and transfusion historyGlanzmann ThrombastheniaAmong the 11 GT patients, 5 patients presented a positive PIFT, suggesting the existence of platelet alloantibodies, while the other 6 patients had a negative PIFT (Table 1). The positive results were confirmed by the MAIPA and PAKLx in 2 of those patients, with the identification of an anti-HLA antibody for one patient (patient number 5 in Table 1) and an anti-HPA antibody for the other one (patient number 6 in Table 1).
Alloimmunization and transfusion history for GT patients.
For the analysis of the transfusion history, the counting of units considered random, pool and apheresis units. All random units were leukodepleted. The 46-year-old patient presenting the anti-HLA antibody received 34 units of platelets (PLTs) (the last transfusion occurred 590 days before the test); 2 units of red blood cells (RBCs) and had previously required the administration of the rFVIIa as prophylaxis for an invasive procedure due to the previous history of immune platelet refractoriness. When tested in the PRA-HLA, this patient sample reacted against 97.69% of the panel. This patient was also alloimmunized for RBC antigens (undetermined cold agglutinin antibody).
The 25-year-old patient presenting the anti-HPA antibody (anti-αIIbβ3) had been transfused with 29 PTLs and 1 RBC, the last PTL transfusion having occurred 2,430 days before the antibody testing. Both patients had type 1 GT and none of them received a transfusion before the age of 5. In regard to transfusion reactions, one of them presented an allergic reaction to the PLTs; one of the patients hadt HCV seropositivity and one was HBsAg positive. Both were HIV and HTLV seronegative.
When analyzing the 3 patients who were positive on the PIFT, but negative on the MAIPA and PAKLx, we observed that they received a mean of 46 PLTs (from 5 to 120) and only one of them received RBCs (16 units). The mean time between the last PLT transfusion and the antibody testing was 1,099 days (from 240 to 2,892) and all three patients were also alloimmunized for RBC antigens. In regard to transfusion reactions, one of them had a previous history of a transfusion-related acute lung injury (TRALI) after the PLT transfusion and one of them was positive for HCV.
Among the 5 patients with a negative PIFT and available data on the previous transfusion, the mean number of PLTs received was 191 (from 3 to 521); the mean of RBCs transfused was 29.2 (from 2 to 73) and 2 of them were also alloimmunized for RBC antigens. Only one of the patients received a transfusion before the age of 5. The mean time between the last PLT transfusion and the PIFT test was 1,848.8 days (from 199 to 4,130) and none of the patients had received the rFVIIa. Considering transfusion reactions, 2 patients presented allergic reactions to the PLT and one also presented one episode of non-hemolytic febrile reaction. One of the patients was seropositive for HCV, HBsAg and HTLV. None of them were HIV positive.
As demonstrated in Table 1, the number of PLT units transfused, as well as the interval between the last PLT transfusion and the sample collection to perform the platelet alloantibody detection, was widely variable in the studied population. However, the presence of alloantibodies not detected by the screening test that would indeed present a clinical problem for patients, but were just below detection at the time of assay, cannot be disregarded.
Bernard-Soulier SyndromeAmong the 4 evaluated patients, 3 had a positive PIFT result. From those 3 patients, only 1 was positive in the MAIPA and PAKLx tests, presenting an anti-HLA antibody. This 51-year-old patient had received 28 PLTs and 17 RBCs. When tested in the PRA-HLA, the patient presented 97.08% of positivity. The interval between the last PLT transfusion and the antibody testing was 67 days. The patient was alloimmunized for RBC antigens (undetermined cold agglutinin antibody) and had a previous history of an allergic reaction to platelet transfusion. This patient was seropositive for HCV and had a previous history of hepatitis B.
In regard to the patients with a positive PIFT and negative MAIPA and PAKLx, one of them was never transfused and one was transfused with only one unit of PLTs 231 days prior to the positive test. Both patients were negative for HCV, HIV, HBsAg and HTLV.
The patient with the negative PIFT was transfused with 6 PLTs and 7 RBCs and the antibody test was performed 143 days after the last platelet transfusion. The patient did not present any transfusion reaction and all the viral serologies were negative. None of the studied patients received Factor VII.
As demonstrated in Table 2, the patient with BSS presenting a platelet alloantibody is the one who received the highest number of platelet transfusions and who also had the shortest interval between the last transfusion and the alloantibody detection testing.
Aloimmunization and transfusion history for BSS patients.
Inherited platelet disorders, such as the Glanzmann Thrombasthenia and Bernard-Soulier Syndrome, are rare hemorrhagic disorders with a frequent need for platelet transfusion and, consequently, the formation of anti-HLA and anti-HPA antibodies in a varying proportion of patients, which can result in platelet refractoriness, directly impacting the clinical management. By implementing three different techniques, we developed an algorithm (Figure 1) for evaluating the presence of alloantibodies in a population of patients with GT and BSS from a reference center in southeastern Brazil and we found a frequency of platelet alloantibodies of 18.1% in the GT patients (n = 2/11 patients - 1 patient with anti-HLA and 1 patient with anti-HPA) and 25% in the BSS patients (n = 1/4 patients - anti-HLA).
Previous historical reviews have described a prevalence of platelet alloantibodies varying from 25% to 49% among GT patients,4,13 which may be anti-HLA or anti-HPA.1 In the international GT registry data, 65/218 (30%) of patients treated with different hemostatic agents developed platelet alloantibodies, of which 47 were against the GPIIb-IIIa and 21 towards the HLA.14,15 The only patient from our cohort who presented an anti-HPA antibody did not have clinical evidence of platelet refractoriness. The presence of the alloantibody in the absence of clinical significance may be explained by previous studies that had already suggested that platelets may be effective, even in patients with platelet alloantibodies, because of long time intervals between exposure to platelets in the GT and perhaps also due to the transient nature of the alloantibodies.16 Another possibility already mentioned in other studies is the presence of “natural” antibodies to HLA antigens, even without transfusion, just like the anti-A and anti-B.17 In our study, the early initiation of the use of platelet transfusion does not seem to have interfered with the formation of alloantibodies, as none of the patients with detected alloantibodies received transfusion before the age of 5.
In the context of the clinical management of bleeding or prophylaxis for invasive procedures in patients with these disorders, the identification of platelet alloantibodies is extremely important, as an important therapeutic strategy for patients with GT is the use of rFVIIa, which is a high-cost medication, with an approved indication only for patients with a previous or current history of platelet refractoriness and identified anti-HPA and / or anti-HLA antibodies in Brazil. We identified 2 patients with anti- HLA antibodies showing reactivity greater than 97% in the PRA-HLA testing. This positivity rate is critical, as it will make it extremely difficult to access a negative crossmatch apheresis platelet, requiring an extensive HLA-typed donor bank to find a compatible donor. In this scenario, the rFVIIa becomes a fundamental therapeutic tool. Since 2014, the rFVIIa had been approved by the FDA for use in the US in patients with GT with refractoriness to platelet transfusions, with or without antibodies for platelets. Similarly, the European label for the rFVIIa has been extended recently to include patients with GT with past or present refractoriness to platelet transfusions, or where platelets are not readily available.18 However, in many other countries, such as Brazil, currently the detection of platelet alloantibodies is still essential for these patients to have access to this therapeutic option.
In order to facilitate, systematize and reduce the cost of identifying these antibodies, through this study we developed the algorithm in Figure 1. From the results of this study, we suggest the application of the PIFT as an initial screening test for the detection of platelet alloantibodies in all patients with GT or BSS, even if they have never been previously transfused. Furthermore, it is of great value in patient management, since in case of a positive result, it is even possible to select a platelet unit to be transfused to the patient if necessary, as at least 4 different platelet samples are used in the methodology of the test. Although the PIFT has a good sensitivity, it does not discriminate between anti-HLA and anti-HPA antibodies.19 After an initial positive result, it is necessary to confirm it using a test of higher specificity, such as the MAIPA or PAKLx, that also allow us to identify which alloantibody is present. In our study, we found a 100% agreement between the results of both techniques, similar to previous studies,11 suggesting that any of them can be used individually. The two methodologies use more laborious techniques and are more expensive, which is why they could be reserved only for initially positive patients in the PIFT test.20
To our knowledge, this is the first description of frequency of alloimmunization in inherited platelet disorders from Latin America. Patients included in the study have been followed in the same outpatient clinic for a long time and by the same medical team. All patient samples were tested in the same reference immunohematology laboratory, using the same techniques, which ensures the reproducibility of the results. The access to different laboratory techniques for the detection of alloantibodies allowed us to evaluate the results looking for the best cost benefit in the process of identifying these antibodies.
As limitations, data on clinical characteristics and bleeding manifestations were collected retrospectively, which limits the availability of detailed data. The time of blood collection for the antibody testing was not related to the moment of the transfusion and the test was performed only once for each patient, without a follow-up. Our study was not able to evaluate the hypothesis of fewer antibodies in patients with type 2 GT caused by the protective effect of some amount of antibody because none of our patients had the type 2. Another major limitation is the small number of patients included, which is justified by the rarity of these disorders.
ConclusionIn conclusion, our findings describe a relatively low frequency of alloimmunization in our population of patients with hereditary platelet disorders. However, we emphasize the extreme importance of identifying these antibodies in the clinical management of these patients, to enable access to the rFVIIa. Following the algorithm we developed, it is possible to perform the detection in steps, maximizing the use of resources and making the best use of each technique and the results they provide. Further studies in a larger number of patients are necessary for a better understanding of antibody frequency and factors that lead to the formation, as well as to define the clinical significance of these alloantibodies.
C.G. helped design the study and collect the patient samples, wrote the manuscript and approved the final version; K.C.Z. collected the patient samples, performed the tests, edited the manuscript and approved the final version; N.D. helped perform the tests and approved the final version of the manuscript; F.M.P. helped collect the patient samples and approved the final version of the manuscript; V.B.O. and M.C.A.V.C helped perform the tests and approved the final version of manuscript; M.R.D. helped perform the tests and approved the final version of the manuscript; A.M.J. edited the manuscript and approved the final version; P.R.V. helped design the study, edited the manuscript and approved the final version, and; C.L.D. and V.R. conceived the study, wrote the manuscript and approved the final version.


