


To describe cytogenetic and molecular abnormalities observed in children and adolescents with acute myeloid leukemia (AML), classify AML according to the World Health Organization (WHO) classifications from 2008 and 2016, and evaluate the prognosis according to clinical characteristics and cytogenetic abnormalities.
MethodsA retrospective longitudinal study was performed on a population of 98 patients with AML, aged up to 16 years, seen in a single hospital from 2004 to 2015.
ResultsAmong the 80 patients for whom it was possible to analyze the karyotype, 78.7% had chromosomal changes, the most frequent being t(15;17)(q22;q21). Of the 86 patients for whom we had cytogenetic or molecular data, making it possible to classify their AML according to the WHO classification, 52.3% belonged to the group with recurrent genetic abnormalities, 22% to the “AML not otherwise specified” group, 18.6% to the group with myelodysplasia-related cytogenetic changes, and 7% to the group with Down syndrome-related leukemia. Five-year overall survival (OS) for the whole group was 49.7%±5.2%. In the univariate and multivariate analyses, patients with myelodysplasia-related cytogenetic changes (OS 28.1%±12.2%) and those with “AML not otherwise specified” (OS 36.1%±11.2%) had an unfavorable prognosis when compared to patients with AML with recurrent genetic abnormalities (OS 71%±5.8%) and patients with Down syndrome-related AML (OS 83%±15.2%, p=0.011).
ConclusionsThe results corroborate the importance of cytogenetic abnormalities as a prognostic factor and indicate the need for cooperative and prospective studies to evaluate the applicability of the WHO classification in the pediatric population.
Over the last 3 decades, there has been a significant improvement in the prognosis of children with acute myeloid leukemia (AML). Survival rates between 60 and 70% have been reported.1,2 These advances are due to improvements in the therapeutic approach during the induction and post-remission phases, treatment stratification according to the risk of relapse, the advancement of supportive therapies, the execution of allogeneic hematopoietic stem cell transplantation in patients with elevated risk of relapse, and the monitoring of minimal residual disease (MRD).1–4
Structural or numerical chromosomal abnormalities can be found in nearly 70% of children with AML.1 Several chromosomal rearrangements have been associated with distinct morphological subgroups and are recognized as important parameters for diagnostic and follow-up purposes, with implications for defining risk groups for relapse and selecting patients who need more intensive treatment.5–7
Therefore, the World Health Organization (WHO) classification for myeloid neoplasms has become quite important in the approach of patients with AML.8 In this classification, clinical criteria and cytogenetic and molecular changes were incorporated, which were associated with the morphological and immunophenotypic characteristics used in the classification recommended by the French–American–British (FAB) cooperative group.8,9 However, the WHO classification was mainly based on the experience with adult patients and there is no consensus on its relevance and applicability for the pediatric population.7 Literature on the use of the WHO 2008 classification for children is scarce7,10,11 and until this manuscript was finished, no studies using the latest WHO classification, published in 2016, had been found in the literature.12
The objective of this study was to contribute to expanding knowledge on AML in the pediatric population, especially regarding cytogenetic findings and the use of the WHO classification.
MethodsA retrospective longitudinal study was performed on a population of patients aged up to 16 years, seen at a university hospital from January 2004 to March 2015, with diagnosis of AML established by myelogram. For the patients who fulfilled these inclusion criteria, the reports of the immunophenotyping, cytogenetic, and molecular biology tests available in the institution files were consulted. The clinical and laboratory data were taken from medical records.
The FAB criteria were used in defining the diagnosis of AML and morphological classification.9 Cytogenetic studies were done at a single laboratory at the Institution. The nomenclature of karyotypes followed the rules of the International System for Cytogenetic Nomenclature (ISCN 1995–2013).
Molecular tests were performed by polymerase chain reaction, using samples of bone marrow or peripheral blood to identify the presence of fusion genes (RUNX1-RUNX1T1, CBFB-MYH11, BCR-ABL, and PML-RARA) and the internal tandem duplication of the FLT3 gene (FLT3-ITD). These tests corresponded to those available at the institution from 2004 to 2015 and were done at a single laboratory at the Institution.
The patients were classified retrospectively according to the last 2 versions of the WHO classification (WHO 2008 and WHO 2016).8,12 The subgroups defined according to this classification were:
- 1)
AML with recurrent genetic abnormalities, with or without association with the FLT3-ITD, which was classified according to the detected abnormality.
- 2)
AML with myelodysplasia-related changes; patients with myelodysplasia-related cytogenetic abnormalities, as specified in the WHO classification, were classified in this group. Patients with complex karyotypes according to the definition of the WHO 2008 and 2016 classifications, namely those with more than 3 unrelated changes which are not included in the subgroup “AML with recurrent genetic changes”, were also included in this subgroup. In the present study, the morphological criteria included in the WHO classifications were not evaluated.
- 3)
AML not otherwise specified (NOS). Patients with normal karyotype and those without molecular tests available or evaluable at diagnosis or who did not present a positive result for the molecular changes tested were included in the NOS group.
- 4)
Down syndrome-related AML.
In the present case series, no patient who fit into the “treatment-related myeloid neoplasms” and “myeloid sarcoma” subgroups was identified.
The institution has adopted different protocols for treating AML over the years. From 2004 to November 2007, the modified BFM-83 protocol was used (GMTLMAI),13 based on the protocol of the Berlin-Frankfurt-Munich (BFM) German cooperative group of 1983.14 As of December 2007, the NOPHO-93 protocol, based on the protocol of the Nordic Society of Pediatric Hematology and Oncology (NOPHO), was adopted.15 Up until August 2007, there was no standardized protocol for the treatment of acute promyelocytic leukemia (APL) and, as recommended in the literature, therapeutic regimens that included all-trans retinoic acid, cytarabine, and anthracycline (daunomycin) were used. As of September 2007, the ICP-LPA/APL2006 protocol was implemented.16
The Kaplan–Meier method was used to estimate overall survival (OS) and event-free survival (EFS). Patients who had not undergone any event (death or relapse) were censored on the date of analysis of the results. Patients who had abandoned treatment or follow-up, without relapse or death, were censored on the date they last visited the institution. The log-rank test was used to compare the survival curves. The Cox model was used for multivariate analysis. The results were presented as risk ratios, with the 95% confidence intervals given. Significance was tested based on the likelihood statistic (Wald test). For all analyses, a value of p<0.05 was considered statistically significant.
The study was approved by the institution's Research Ethics Committee (CAAE Opinion-31579014.3.0000.5149). Patients who were still under follow-up at the institution were admitted into the study after providing signed informed consent.
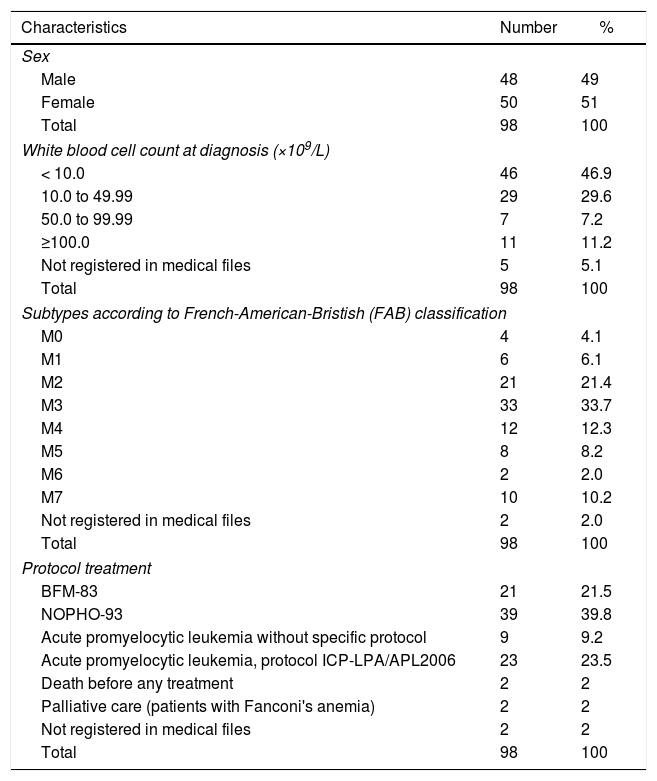
ResultsDuring the period evaluated, 99 patients were diagnosed with AML. A single patient who came from another institution was excluded from the study because treatment had already been initiated and 2 morphological tests had had discordant results. The age at diagnosis ranged from 2 months to 16 years (median, 7.6 years). Initial leukocyte count ranged from 740/mm3 to 218,000/mm3 (median, 10,780cells/mm3). Six patients had Down syndrome. The other characteristics of the case series are included in Table 1.
Characteristics of 98 children and adolescents with acute myeloid leukemia.
| Characteristics | Number | % |
|---|---|---|
| Sex | ||
| Male | 48 | 49 |
| Female | 50 | 51 |
| Total | 98 | 100 |
| White blood cell count at diagnosis (×109/L) | ||
| < 10.0 | 46 | 46.9 |
| 10.0 to 49.99 | 29 | 29.6 |
| 50.0 to 99.99 | 7 | 7.2 |
| ≥100.0 | 11 | 11.2 |
| Not registered in medical files | 5 | 5.1 |
| Total | 98 | 100 |
| Subtypes according to French-American-Bristish (FAB) classification | ||
| M0 | 4 | 4.1 |
| M1 | 6 | 6.1 |
| M2 | 21 | 21.4 |
| M3 | 33 | 33.7 |
| M4 | 12 | 12.3 |
| M5 | 8 | 8.2 |
| M6 | 2 | 2.0 |
| M7 | 10 | 10.2 |
| Not registered in medical files | 2 | 2.0 |
| Total | 98 | 100 |
| Protocol treatment | ||
| BFM-83 | 21 | 21.5 |
| NOPHO-93 | 39 | 39.8 |
| Acute promyelocytic leukemia without specific protocol | 9 | 9.2 |
| Acute promyelocytic leukemia, protocol ICP-LPA/APL2006 | 23 | 23.5 |
| Death before any treatment | 2 | 2 |
| Palliative care (patients with Fanconi's anemia) | 2 | 2 |
| Not registered in medical files | 2 | 2 |
| Total | 98 | 100 |
Regarding immunophenotyping by flow cytometry, results were obtained in 86 of the 98 cases, corroborating the cytomorphologic diagnosis of AML in all of them.
In 90 patients, cytogenetic results were obtained at diagnosis. In 10 of them (11.1%), metaphase was not observed during chromosomal analysis. Thus, the karyotype could only be evaluated in 80 of the patients included in the study: 17 (21.3%) presented normal karyotype and 63 (78.6%) presented chromosomal aberrations. Of these 63 patients, 17 (27%) presented a complex karyotype (e.g. three or more aberrations) and 46 (73%) presented one aberration (38 cases) or two aberrations (8 cases) in their karyotype (Table 2).
Cytogenetic and molecular classification according to the World Health Organization (WHO) in 86 children and adolescents with acute myeloid leukemia (AML).
| Characteristics | Number | % |
|---|---|---|
| Cytogenetic alterations | ||
| Normal karyotype | 17 | 21.3 |
| t(15;17)(q22;q21) | 21 | 26.2 |
| t(8;21)(q22;q22) | 7 | 8.8 |
| inv(16)(p13.1q22) or t(16;16)(p13.1;q22) | 2 | 2.5 |
| t(9;11)(p22;q23) | 2 | 2.5 |
| AML (megakaryoblastic) with t(1;22)(p13;q13) | 2 | 2.5 |
| AML with myelodysplasia-related changes: -5/del(5q), -7/del(7q), del(11q), del(12p)/t(12p), and del(13q) | 13 | 16.2 |
| Other chromosomic abnormalities | 16 | 20.0 |
| Totala | 80 | 100 |
| Fusion genes and specific mutations | ||
| PML-RARA | 25 | 31.6 |
| PML-RARA and FLT3-ITD | 2 | 2.5 |
| CBFB-MYH11 | 3 | 3.8 |
| AML1-ETO (RUNX1-RUNX1T) | 5 | 6.3 |
| BCR-ABL | 0 | 0 |
| FLT3-ITD (as the single abnormality) | 3 | 3.8 |
| Negative tests for the above-mentioned abnormalities | 41 | 52.0 |
| Totalb | 79 | 100 |
| WHO classification | ||
| AML with recurrent genetic abnormalities | 45 | 52.3 |
| t(8;21)(q22;q22)/RUNX1-RUNX1T | 9 | |
| inv(16)(p13.1q22)/CBFB-MYH1 | 3 | 18.6 |
| t(15;17)(q22;q21)/PML-RARA | 29 | |
| t(9;11)(p22;q23) | 2 | 22 |
| AML (megakaryoblastic) with t(1;22)(p13;q13) | 2 | |
| AML with myelodysplasia-related changes | 16 | 7 |
| -7/del(7q) | 4 | |
| -5/del(5q) | 2 | 100 |
| del(11q) | 5 | |
| -13/del(13q) | 1 | |
| del(12p) or t(12p) | 1 | |
| AML secondary to myelodysplastic syndrome | 1 | |
| Complex karyotype | 2 | |
| AML not otherwise specified | 19 | |
| Normal karyotype | 11 | |
| Other abnormalities | 8 | |
| Myeloid leukemia associated with Down syndrome | 6 | |
| Normal karyotype (except for the constitutional abnormality) | 1 | |
| Numerical chromosomal abnormalityc | 2 | |
| Structural chromosomal abnormalityd | 1 | |
| No metaphases for cytogenetic study | 1 | |
| Not registered in medical file | 1 | |
| Totale | 86 | |
Additionally, in 10 patients metaphases were not present for cytogenetic analysis and in 8 patients cytogenetic restults were not registered in medical files.
Additionally, in 16 patients molecular studies were not requested at diagnosis and in three patients DNA/cDNA amplification was not successful.
Regarding the molecular study, out of a total of 98 patients, 16 (16.3%) had had no molecular test at diagnosis. Of the 82 patients tested, 3 were considered non-evaluable, as DNA/cDNA amplification could not be carried out. Results were therefore obtained for 79 (80.6%) patients, and of these, 38 were positive for the gene changes tested (Table 2).
For 86 of the 98 cases included in this study, it was possible to obtain cytogenetic or molecular data that allowed for classification according to the WHO criteria. There was no difference in the classification of the 86 patients when using either of the WHO versions from 2008 or 2016, so the nomenclature was unified for the WHO classification (Table 2).
In total, 14 patients met the WHO criteria for the definition of complex karyotype. Of these, 13 were categorized into the group “AML with myelodysplasia-related changes” and 1 patient was categorized as having Down syndrome-related AML. Three patients who had complex karyotypes but also had some recurrent genetic abnormality, namely t(8;21)(q22;q22) in 2 cases and t(15;17) (q22;q21) in 1 case, were categorized into the group with recurrent cytogenetic changes.
A total of 51 deaths were observed: 10 occurred in the patients in remission; 6 in patients undergoing treatment according to the NOPHO protocol, corresponding to 15.3% of the patients treated using this protocol; 3 in patients submitted to the modified BFM protocol, corresponding to 14.2% of the patients treated using this protocol; and 1 in a patient with APL, treated with the ICP-LPA/APL2006 protocol.
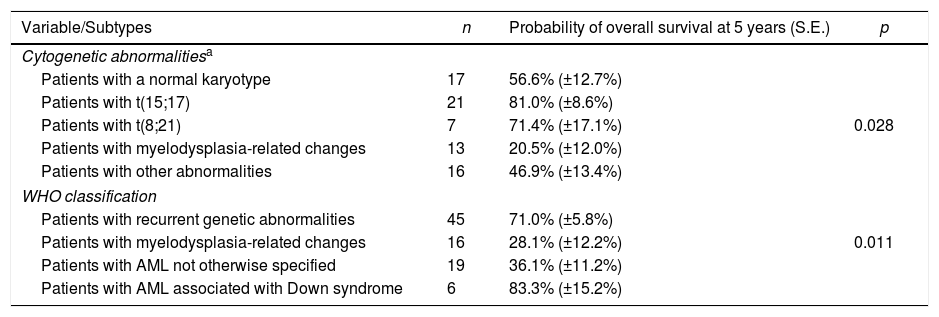
The 5-year OS for all patients was 49.7%±5.2%. For the OS analysis according to cytogenetic changes, as described in Table 3, recurrent cytogenetic changes that were observed in fewer than 5 patients were excluded. The OS, according to the WHO classification, was also recorded in Table 3 and is represented in Figure 1.
Overall survival according to cytogenetic abnormalities or to the World Health Organization (WHO) classification of myeloid neoplasias.
| Variable/Subtypes | n | Probability of overall survival at 5 years (S.E.) | p |
|---|---|---|---|
| Cytogenetic abnormalitiesa | |||
| Patients with a normal karyotype | 17 | 56.6% (±12.7%) | |
| Patients with t(15;17) | 21 | 81.0% (±8.6%) | |
| Patients with t(8;21) | 7 | 71.4% (±17.1%) | 0.028 |
| Patients with myelodysplasia-related changes | 13 | 20.5% (±12.0%) | |
| Patients with other abnormalities | 16 | 46.9% (±13.4%) | |
| WHO classification | |||
| Patients with recurrent genetic abnormalities | 45 | 71.0% (±5.8%) | |
| Patients with myelodysplasia-related changes | 16 | 28.1% (±12.2%) | 0.011 |
| Patients with AML not otherwise specified | 19 | 36.1% (±11.2%) | |
| Patients with AML associated with Down syndrome | 6 | 83.3% (±15.2%) | |
Three recurrent genetic abnormalities [inv(16) or t(16;16), t(9;11), and t(1;22)] were excluded from this analysis because only two patients in each subtype were detected (see Table 2).
, according to World Health Organization (WHO) classification. Kaplan–Meier curves were compared with the logrank test (p=0.011). WHO Down: AML associated with Down syndrome; WHO recurring abnormalities: AML with recurrent genetic abnormalities; WHO NOS: AML not otherwise specified; WHO myelodisplasia-related: AML with myelodysplasia-related changes.")
Estimated probability of overall survival for 86 children with acute myeloid leukemia (AML), according to World Health Organization (WHO) classification. Kaplan–Meier curves were compared with the logrank test (p=0.011). WHO Down: AML associated with Down syndrome; WHO recurring abnormalities: AML with recurrent genetic abnormalities; WHO NOS: AML not otherwise specified; WHO myelodisplasia-related: AML with myelodysplasia-related changes.
Regarding the treatment protocol used, the 5-year OS was 33.3%±10.3% for patients treated with the modified BFM protocol and 40.7%±7.9% for patients treated with the NOPHO protocol (p=0.86).
Patients diagnosed with APL were evaluated separately. The 5-year OS was 44.4%±16.6% for patients treated in the period prior to August 2007, when there was no standardized protocol, and 95.7%±4.3% for the patients treated with the ICP-LPA/APL2006 protocol (p=0.02).
Regarding the leukocyte count at diagnosis, the 86 patients classified according to the WHO classification were divided into 2 groups. The 5-year OS was 61.9%±5.9% for the group of patients with initial leukocyte count below 100,000/mm3 and 25%±15.3% for the group with a count greater than 100,000/mm3 (p=0.01).
The WHO classification and leukocyte count at diagnosis were included in the final multivariate model. The AML patients with myelodysplasia-related changes had a 2.97-fold greater chance of death than patients with recurrent abnormalities (95% CI: 1.33–6.64; p=0.008). Patients of the NOS category, in relation to the baseline category, presented a 2.22-fold greater chance of death, with a tendency to statistical significance (95% CI: 0.94–4.47; p=0.069). Patients with Down syndrome had a 1.7-fold lower chance of death than the baseline category, but without statistical significance (95% CI: 0.075–4.47; p=0.6). The effect of the leukocyte count (above or below 100,000/mm3) was not independent (95% CI: 0.91–5.6, p=0.078) from the WHO classification, although there was a tendency toward said independence.
Data collection for the EFS calculation was only possible for patients diagnosed with APL treated by the ICP-LPA/APL2006 protocol, since it was difficult to define accurately the time of relapse in several patients in the other groups. Five-year EFS for these patients was 79.3%±9.6%. The only death observed in this group, which had already been reported, occurred in a patient who was in remission after the consolidation phase, and this was due to cardiogenic shock. Four relapses were observed. All these patients were alive when the data were analyzed.
DiscussionThe case series of this study represents the 12-year experience of a Brazilian institution and includes only children and adolescents. Similar studies with more representative case series were mostly multicentric and also included patients older than 18 years.7,10,11,17 The frequency of cytogenetic abnormalities observed was similar to that reported in the literature describing the presence of chromosomal rearrangements in 70%–80% of the cases of pediatric AML.1,18 The high percentage of APL cases, detected by morphological, cytogenetic, or molecular findings, was compatible with that described in the literature for populations of Latin origin.18–21
The frequency of normal karyotypes was also similar to that reported in the literature.5 The molecular study identified the PML-RARA fusion gene in 5 patients with normal karyotype, making it possible to confirm the diagnosis of APL and its classification according to the WHO criteria. It should also be highlighted that patients with normal karyotype may present molecular changes, such as mutations in the FLT3-ITD, MLL-PTD, or NPM1. Of these changes, only the presence of a mutation in the FLT3-ITD was investigated in this case series and only in some of the patients. The FLT3-ITD, identified in nearly 10% to 15% of pediatric patients with AML,22 was considered an unfavorable prognostic factor in several studies.18,20,23
Regarding the WHO classification, it should be taken into consideration that it was based primarily on data from adult patient experiences and includes most, but not all, of the subgroups of cytogenetic changes specific to the pediatric age range.7,10 In the present study, the distribution of patients, according to the WHO classification, was similar to that observed by Davis et al.7 and Sandahl et al.,10 who also identified a predominance of patients in the group with recurrent genetic abnormalities (52.3%, 45.9%, and 41%, respectively). The slightly higher frequency of recurrent genetic abnormalities in the present case series may have been influenced by the higher number of patients with t(15;17)(q22;q21).
The NOS subtype was the second most frequent (22%), but due to its heterogeneity, it is difficult to draw a comparison with the frequency reported in other studies (Davis et al.,7 16.2% and Sandahl et al.,10 44%). Although conventional cytogenetics can reliably detect most abnormalities in the pediatric AML population, the limitations of the present study were related to molecular testing and non-use of complementary techniques, such as fluorescence in situ hybridization (FISH), which may be useful in characterizing previously unidentified chromosomal abnormalities or those that are more “subtle”, such as inv(16), t(11q23), and t(7;12), did not allow for better characterization of the group of patients included in the NOS category. If these methods had been available at the institution during the period studied, some of these patients would possibly have been classified into other categories.5,24 Furthermore, the review of morphological findings, which was not performed in this study, could have helped to identify dysplasias in patients without cytogenetic abnormalities, making it possible to categorize them into the group of patients with myelodysplasia-related abnormalities, reducing the number of patients classified as NOS.7,10,11
The WHO 2008 and 2016 classifications incorporated modifications that allowed for a greater number of pediatric patients to be classified into the category of AML with myelodysplasia-related abnormalities.7 However, this category is still controversial in studies with pediatric populations.11 In the present case series, 18.6% of patients were included in this category, a frequency that was slightly higher than that observed by Davis et al. (16%2)7 and Sandahl et al. (14.7%2).10 A Japanese study conducted specifically for the evaluation of this category found that 21% of the cases studied should be included in this group, emphasizing the need for cooperative studies with a greater number of cases to evaluate the real incidence of this subgroup in the pediatric population. These authors also pointed out that, in this age group, myelodysplasia-related cytogenetic changes were not necessarily associated with morphological changes.11 Another important consideration is the fact that cytogenetic changes with very different implications in the prognosis of childhood AML were included in this category.10,11
The OS observed in the present study was similar to that found in a recent Brazilian study,21 but lower than the reports in the international literature.1,16 Some hypotheses can be considered to explain this result, and specifically the high percentage of deaths of patients in remission (19.6%). This value was much higher than that described in the literature, where it was reported that 3% to 10% of AML patients died due to complications of the disease, such as fungal or bacterial infections, or adverse effects related to the intensity of the therapeutic protocols.1,17,25
It should also be highlighted that in the present study, only patients diagnosed with APL, treated according to the ICP-APL protocol, had planned treatment with stratification of patients at risk for relapse and monitoring for early identification of molecular relapse or absence of molecular remission. For the other AML subtypes, the institution had yet to implement a protocol with risk stratification and only had minimal residual disease monitoring, different from that which is recommended in the literature.3,16
When the OS was evaluated according to the identification of cytogenetic abnormalities, the favorable prognosis of patients with t(15;17)26 and t(8;21) was confirmed.7,25 The group of patients with myelodysplasia-related changes, which included abnormalities associated with unfavorable prognosis, such as chromosome 7 monosomy and abnormalities of 5q, presented a lower 5-year OS, as expected.17
Regarding the survival analyses of patients according to the WHO classification, it was observed in the univariate analysis that the group of patients with recurrent genetic abnormalities and those with Down Syndrome-related AML presented favorable prognoses, with results similar to those demonstrated in other studies.7,9,27 The multivariate analysis confirmed the good prognosis of patients with recurrent genetic alterations and Down syndrome-related AML, and the unfavorable prognosis of patients with myelodysplasia-related changes and of the NOS category. In the univariate analysis, but not in the multivariate analysis, lower OS was also observed for patients with an initial leukocyte count above 100,000/mm3. According to several studies, an extremely high blast count at diagnosis was associated with an increased risk of early death and absence of response to treatment, but was not necessarily associated with worse disease-free survival.1,11,28
The detailed evaluation of the results regarding the different treatment protocols used in this study was not possible due to the limitations already mentioned. However, it was possible to observe that the OS of patients treated according to the NOPHO protocol (40.7%) was lower than that reported in the literature. The OS reported in a study by Lie et al.15 for the NOPHO-AML-93 protocol, on which the therapeutic scheme used in the present case series was based, was 65% at 5 years. The OS achieved with the NOPHO protocol was superior to that obtained with the modified BFM-83 protocol,13 but with no statistical significance. These results demonstrated that there is a need to reassess the strategies adopted at the institution to treat patients with AML, especially with respect to reducing the high frequency of deaths among patients in remission.
Regarding patients with APL, there was a significant improvement in the OS with the introduction of the ICP-LPA protocol. The results obtained with this protocol, in relation to the OS and EFS, were similar to the results described in the literature15,26 and suggested that the adoption of a unified therapeutic protocol may have contributed to the marked increase in survival rates in this group of patients.
ConclusionsFor proper interpretation of the conclusions of this study, we should consider the limitations regarding its retrospective nature, which contributed to difficulties in obtaining certain data, and to the relatively small size of the case series. Limitations related to the study of molecular changes and the non-use of complementary techniques, such as the FISH technique, should also be noted.5,23
Even with the limitations presented, the results corroborate the importance of cytogenetic abnormalities as a prognostic factor for pediatric patients with AML and indicate the need for cooperative and prospective studies in order to better understand the characteristics of the disease and to evaluate the applicability of the WHO classification in the pediatric population to improve the treatment of these patients and to increase their chances of cure.
Conflicts of interestThe authors declare no conflicts of interest.



