

The classical BCR-ABL1-negative myeloproliferative neoplasms (MPNs) are Polycythemia Vera (PV), Essential Thrombocythemia (ET) and Primary Myelofibrosis (PMF). In developing countries, there are few reports that truly reveal the clinical setting of these patients. Therefore, we aimed to characterize a single center MPN population with a special focus on the correct diagnosis based on the recent review of the WHO criteria for the diagnosis of myeloid neoplasms.
MethodsThis retrospective study analyzed data from medical records of patients with classical BCR-ABL1-negative MPNs diagnosed from January 1997 to October 2017 and followed at the University Hospital of Ribeirão Preto Medical School.
ResultsA total of 162 patients were assessed, 61 with PV, 50 with ET, and 51 with PMF. The mutational status analysis revealed that 113 (69.3%) harbored the JAK2V617F mutation, 23 (14.1%), the CALR mutation, and 12 (7.4%) had a triple-negative status. None of the patients were found to have mutations on the thrombopoietin receptor gene (MPL), including some ET and PMF patients who were not tested. Among the PV patients, 57 (93.5%) were positive for the JAK2V617F mutation, one (1.6%) presented an in-frame deletion JAK2 exon 12 mutation and one (1.6%) presented a missense JAK2 exon 9 mutation, not previously described. The overall survival was lower in the triple-negative patients with PMF, when compared to the JAK2V617F or CALR-mutated (p = 0.002).
ConclusionThe frequency of somatic mutations and survival in our cohort, stratified according to the respective disease, was consistent with the literature data, despite some limitations. Further prospective epidemiological studies of MPN cohorts are encouraged in developing countries.
The classical BCR-ABL1-negative myeloproliferative neoplasms (MPNs) are a group of disorders characterized by constitutive activation of the physiologic Janus kinase signal transducer and activator of transcription (JAK-STAT) signal-transduction pathways responsible for hematopoiesis1 and consequently, excessive production of terminally differentiated blood cells that are fully functional.2 According to the new 2016 World Health Organization (WHO) classification of Hematological Malignancies, they can be classified into three classical entities: Polycythemia Vera (PV), Essential Thrombocythemia (ET) and Primary Myelofibrosis (PMF). The latter is further divided into prefibrotic and overt fibrotic stages.3 These disorders present a variable increase in blood cell counts, that may include either erythrocytosis, leukocytosis or thrombocytosis, and are clinically characterized by the presence of constitutional symptoms, thrombotic events, bleeding episodes, and increased risk of progression to acute myeloid leukemia.
Three main genes are responsible for clonal expansion of the hematopoietic stem cell and consequently single or multilineage hyperplasia.2 The first discovered driver mutation consists of a substitution of valine for phenylalanine at position 617 in exon 14 of the Janus kinase (JAK2) gene (JAK2V617F).4,5 Subsequently, other mutations were found in exon 9 of the calreticulin gene (CALR)6,7 and exon 10 of the thrombopoietin receptor gene (MPL).8,9 Furthermore, in JAK2V617F-negative PV cases, several mutations of JAK2 exon 12 have been found.10,11
The phenotype of these disorders is associated with these three main driver and mutually exclusive mutations, with JAK2V617F occurring in approximately 97% of the PV cases and at least 50% of ET and PMF, whereas CALR and MPL are mutated in 25–35% of ET and PMF patients.2 The absence of these mutations in MPNs characterizes a triple-negative status that is associated with lower overall survival rates and higher incidence of leukemic transformation.12,13
In developing countries, there are a few reports that truly reveal the clinical setting of patients with myeloproliferative neoplasms. In addition, the recent update of the new diagnostic criteria that emphasize the relevance of histopathologic and molecular features of the classical MPNs has been only recently incorporated in the clinical practice. Therefore, we aimed to improve the knowledge on the clinical profile of a Brazilian MPN subpopulation, whose diagnosis has been confirmed according to the optimized 2016 WHO criteria. This information may contribute to the understanding of the real needs of such patients regarding diagnosis, risk stratification and management, that will potentially improve the quality of care for MPN.
Materials and methodsPatient demographicsWe conducted an epidemiological retrospective study of patients with BCR-ABL1-negative MPNs diagnosed from January 1997 to October 2017 and followed up at the University Hospital of Ribeirao Preto Medical School.
The main objectives were to evaluate the clinical and mutational profile, as well as the overall survival, of a group of patients with classical BCR-ABL1-negative MPNs regarding the respective phenotypic mutation, with a special focus on the correct diagnosis, based on the recent review of the WHO criteria for myeloid neoplasm diagnosis.
The patients were identified in the database of the specialized outpatient clinic. Data considering clinical, demographic and mutational characteristics, as well as laboratory tests, were collected and reviewed from their medical records.
Inclusion and exclusion criteriaPatients diagnosed with PV, ET or PMF were included according to the new 2016 WHO classification. A total of 27 patients were excluded because of incomplete data in medical records, making it impossible to confirm the diagnosis.
Molecular characterizationThe mutational status was assessed using a DNA-based Real Time PCR (Polymerase Chain Reaction) technique for the JAK2V617F mutation and a quantitative RT-PCR (Reverse Transcription PCR) technique for the exclusion of the BCR-ABL1 fusion transcript. The DNA Sanger sequencing was used for the JAK2 mutational screening beyond the exon 14 V617F mutation and/or for MPL mutations. The CALR exon 9 mutations were assessed by the Fragment Analysis Genotyping in DNA samples from mononuclear cells obtained from peripheral blood and/or bone marrow.
Statistical analysisStatistical analyses were performed using the GraphPad Instat 5 (GraphPad Software, Inc., San. Diego, CA, USA). For comparisons among the three groups, the chi-square test was used for categorical factors and the Kruskal–Wallis test was used for measured factors. For survival analyses, the overall survival (OS) was defined as the time (in years) between the date of sampling and the date of death (for deceased patients) or the last follow-up (for censored patients), which was estimated by the Kaplan–Meyer curves and compared by the Log-rank test. A p-value <0.05 was considered statistically significant.
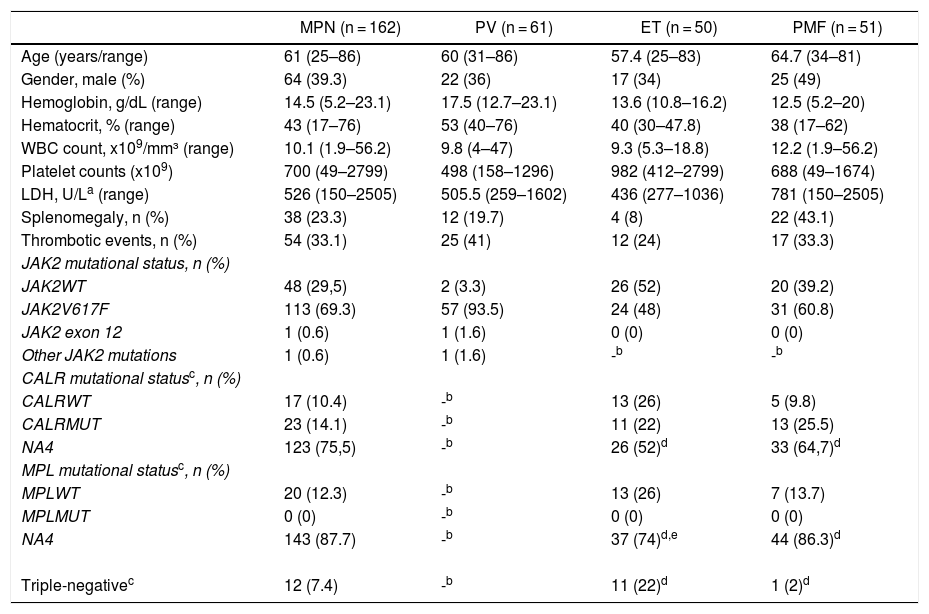
ResultsA total of 162 patients with classical BCR-ABL1-negative MPNs were assessed, 61 with PV, 50 with ET, and 51 with PMF [Table 1]. The median follow-up (minimum-maximum) in years was 4.37 (0.27–20.52) for PV patients, 4.06 (0.06–17.14) for ET patients, and 4.18 (0.19–13.18) for PMF patients. The mean age for all MPNs was 61 years (range of 25–86), with the average age of PMF being slightly higher than the average age of PV or ET patients (64.8 years, 60 years, and 57.4 years, respectively). The male:female ratio was 1:1.8 for PV patients, 1:1.9 for ET patients, and 1:1 for PMF patients.
Demographic features.
| MPN (n = 162) | PV (n = 61) | ET (n = 50) | PMF (n = 51) | |
|---|---|---|---|---|
| Age (years/range) | 61 (25–86) | 60 (31–86) | 57.4 (25–83) | 64.7 (34–81) |
| Gender, male (%) | 64 (39.3) | 22 (36) | 17 (34) | 25 (49) |
| Hemoglobin, g/dL (range) | 14.5 (5.2–23.1) | 17.5 (12.7–23.1) | 13.6 (10.8–16.2) | 12.5 (5.2–20) |
| Hematocrit, % (range) | 43 (17–76) | 53 (40–76) | 40 (30–47.8) | 38 (17–62) |
| WBC count, x109/mm³ (range) | 10.1 (1.9–56.2) | 9.8 (4–47) | 9.3 (5.3–18.8) | 12.2 (1.9–56.2) |
| Platelet counts (x109) | 700 (49–2799) | 498 (158–1296) | 982 (412–2799) | 688 (49–1674) |
| LDH, U/La (range) | 526 (150–2505) | 505.5 (259–1602) | 436 (277–1036) | 781 (150–2505) |
| Splenomegaly, n (%) | 38 (23.3) | 12 (19.7) | 4 (8) | 22 (43.1) |
| Thrombotic events, n (%) | 54 (33.1) | 25 (41) | 12 (24) | 17 (33.3) |
| JAK2 mutational status, n (%) | ||||
| JAK2WT | 48 (29,5) | 2 (3.3) | 26 (52) | 20 (39.2) |
| JAK2V617F | 113 (69.3) | 57 (93.5) | 24 (48) | 31 (60.8) |
| JAK2 exon 12 | 1 (0.6) | 1 (1.6) | 0 (0) | 0 (0) |
| Other JAK2 mutations | 1 (0.6) | 1 (1.6) | -b | -b |
| CALR mutational statusc, n (%) | ||||
| CALRWT | 17 (10.4) | -b | 13 (26) | 5 (9.8) |
| CALRMUT | 23 (14.1) | -b | 11 (22) | 13 (25.5) |
| NA4 | 123 (75,5) | -b | 26 (52)d | 33 (64,7)d |
| MPL mutational statusc, n (%) | ||||
| MPLWT | 20 (12.3) | -b | 13 (26) | 7 (13.7) |
| MPLMUT | 0 (0) | -b | 0 (0) | 0 (0) |
| NA4 | 143 (87.7) | -b | 37 (74)d,e | 44 (86.3)d |
| Triple-negativec | 12 (7.4) | -b | 11 (22)d | 1 (2)d |
MPN, myeloproliferative neoplasm; ET, essential thrombocythemia; PV, polycythemia vera; PMF, primary myelofibrosis; WBC, white blood cell count; JAK2, Janus kinase 2; CALR, Calreticulin; MPL, thrombopoietin receptor; WT, wild-type; MUT, mutated; LDH, lactate dehydrogenase; NA, not available.
As expected, regarding complete blood count parameters, we found that the values of the hemoglobin and hematocrit were higher in PV patients, when compared to ET or PMF patients, with a median hemoglobin level of 17.5 g/dL in PV, 13.6 g/dL in ET and 12.5 g/dL in PMF. White blood count levels were higher in PMF patients (median of 12.2 × 109/L), when compared to PV patients (median of 9.8 × 109/L) or ET patients (median of 9.3 × 109/L). Platelet counts were higher in ET patients (median of 982 × 109/L), when compared to PV patients (median of 498 × 109/L) or PMF patients (median of 688 × 109/L). Lactate dehydrogenase (LDH; normal range: 230–460 U/L) levels were higher in PMF patients (median of 781 U/L), with median values in PV and ET patients of 505 U/L and 436 U/L, respectively.
When clinical characteristics were assessed in our population, splenomegaly was observed in 22 (43.1%) of the patients with PMF, 12 (19.7%) with PV, and 4 (8%) with ET. Of note, a total of 54 (33.1%) patients with MPN experienced some thrombotic event, of which 25 (41%) had PV, 12 (24%) ET, and 17 (33.3%) PMF. The main thrombotic events were stroke, acute myocardial infarction, portal vein thrombosis and peripheral arterial thrombosis.
The analysis of the mutational status revealed that a total of 113 (69.3%) patients with MPN harbored the JAK2V617F mutation, 23 (14.1%) harbored the CALR mutation and 12 (7.4%) presented a triple-negative status (negative for JAK2V617F, CALR and MPL). None of our patients were found to have the exon 10 MPL mutation. Among all PV patients, 57 (93.5%) were positive for the JAK2V617F mutation, one (1.6%) presented an inframe deletion JAK2 exon 12 mutation [p.Phe537_Lys539delinsLeu (c.1611_1616delTCACAA)] and 3 (4.8%) were not found to have mutations on exons 12 or 14. Among the latter, we identified a missense JAK2 exon 9 mutation [p.Leu393Val (c.1177C > G)], not previously described and whose pathogenicity is uncertain, when further sequencing of the JAK2 gene was performed. In the ET group, 24 (48%) were positive for the JAK2V617F mutation, 11 (22%) for the CALR mutation, and 11 (22%) were triple-negative for the three driver mutations. Two patients with the ET and JAK2WT/CALRWT status were not tested for MPL mutations. Among the PMF patients, 31 (60.8%) were positive for the JAK2V617F mutation, 13 (25.5%) for the CALR mutation, and 1 (2%) was a triple-negative. Four patients with PMF and JAK2WT/CALRWT were not tested for MPL mutations [Fig. 1].
, Essential Thrombocythemia (B) and Primary Myelofibrosis (C) patients.")
Regarding the overall survival (OS), patients in the PMF group presenting a triple-negative status were found to have a poorer OS, when compared to patients with the JAK2V617F or CALR mutations (p = 0.002). By contrast, no significant difference in the OS was detected in patients with PV or ET, regardless of their mutational status.
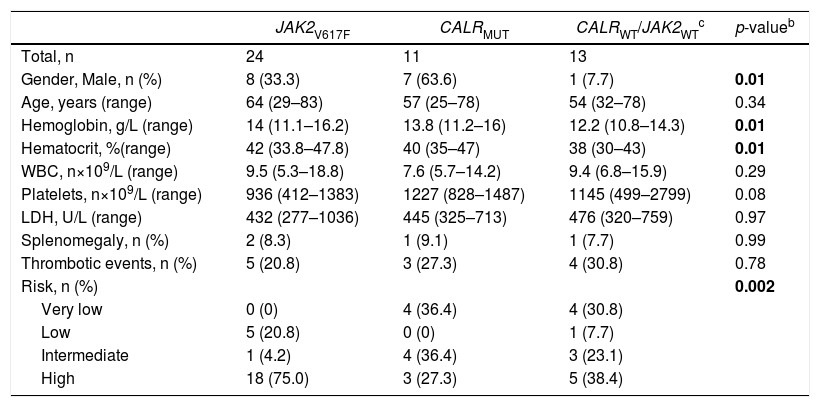
The stratification of ET patients according to the mutational status indicated significant clinical and laboratorial differences regarding gender, hemoglobin/hematocrit and disease risk [Table 2]. More women presented the JAK2V617F mutation, as compared to men, whereas more men presented the CALR mutation, when compared to women (p = 0.01). Most of the patients with wild-type CALR or JAK2 (JAK2wt/CALRwt) were women (p = 0.01). Higher levels of hemoglobin and hematocrit were seen in JAK2V617F-mutated patients, as compared to lower levels detected in the JAK2wt/CALRwt patients (p = 0.01). Higher levels of platelets were observed in patients with the CALR mutation, although this finding did not reach statistical significance (p = 0.08). Lastly, high-risk patients presented the JAK2V617F mutation more frequently, whereas low-risk patients most commonly harbored the JAK2 in its wild-type form (p = 0.002).
Clinical and laboratory features of ET patients, stratified according to phenotypic mutation status (n = 48)a.
| JAK2V617F | CALRMUT | CALRWT/JAK2WTc | p-valueb | |
|---|---|---|---|---|
| Total, n | 24 | 11 | 13 | |
| Gender, Male, n (%) | 8 (33.3) | 7 (63.6) | 1 (7.7) | 0.01 |
| Age, years (range) | 64 (29–83) | 57 (25–78) | 54 (32–78) | 0.34 |
| Hemoglobin, g/L (range) | 14 (11.1–16.2) | 13.8 (11.2–16) | 12.2 (10.8–14.3) | 0.01 |
| Hematocrit, %(range) | 42 (33.8–47.8) | 40 (35–47) | 38 (30–43) | 0.01 |
| WBC, n×109/L (range) | 9.5 (5.3–18.8) | 7.6 (5.7–14.2) | 9.4 (6.8–15.9) | 0.29 |
| Platelets, n×109/L (range) | 936 (412–1383) | 1227 (828–1487) | 1145 (499–2799) | 0.08 |
| LDH, U/L (range) | 432 (277–1036) | 445 (325–713) | 476 (320–759) | 0.97 |
| Splenomegaly, n (%) | 2 (8.3) | 1 (9.1) | 1 (7.7) | 0.99 |
| Thrombotic events, n (%) | 5 (20.8) | 3 (27.3) | 4 (30.8) | 0.78 |
| Risk, n (%) | 0.002 | |||
| Very low | 0 (0) | 4 (36.4) | 4 (30.8) | |
| Low | 5 (20.8) | 0 (0) | 1 (7.7) | |
| Intermediate | 1 (4.2) | 4 (36.4) | 3 (23.1) | |
| High | 18 (75.0) | 3 (27.3) | 5 (38.4) |
ET, essential thrombocythemia); WBC, white blood cell count); JAK2, Janus kinase 2; CALR, Calreticulin; MPL, thrombopoietin receptor; WT, wild-type; MUT, mutated; LDH, lactate dehydrogenase.
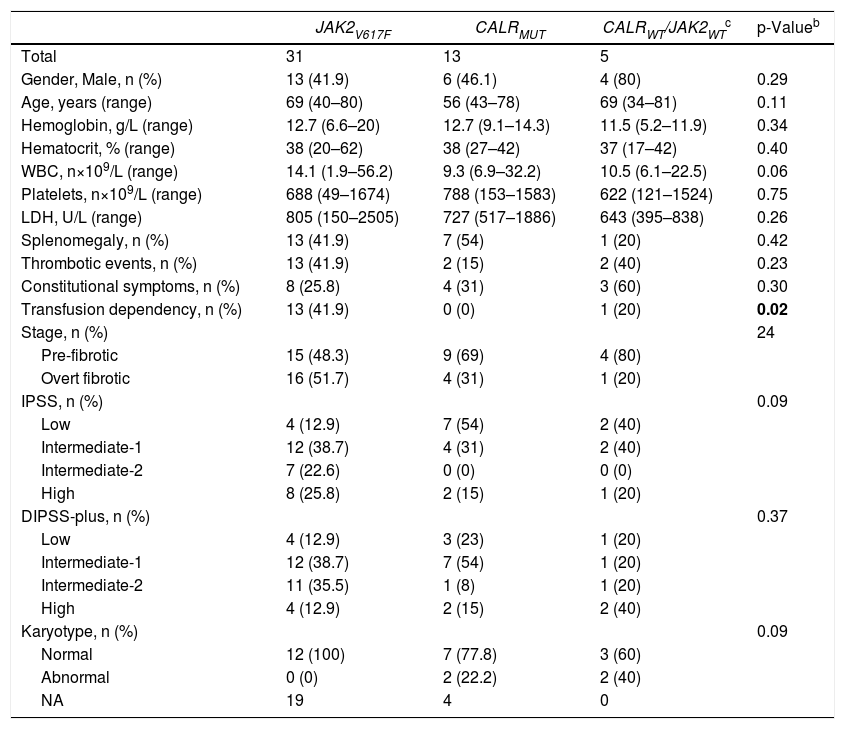
Information about PMF patients according to their mutational status are displayed in Table 3. Of note, patients harboring JAK2V617F mutation had more transfusion dependency (p = 0.02). On the other hand, patients harboring CALR mutation were younger, had lower white blood counts, and low risk disease according to IPSS, although these findings did not reach statistical significance. In contrast, the majority of patients with high risk DIPSS-plus score and abnormal karyotype were JAK2wt/CALRwt or triple-negative, but still with no statistical significance in our population.
Clinical and laboratory features of PMF patients, stratified according to phenotypic mutation status (n = 49)a.
| JAK2V617F | CALRMUT | CALRWT/JAK2WTc | p-Valueb | |
|---|---|---|---|---|
| Total | 31 | 13 | 5 | |
| Gender, Male, n (%) | 13 (41.9) | 6 (46.1) | 4 (80) | 0.29 |
| Age, years (range) | 69 (40–80) | 56 (43–78) | 69 (34–81) | 0.11 |
| Hemoglobin, g/L (range) | 12.7 (6.6–20) | 12.7 (9.1–14.3) | 11.5 (5.2–11.9) | 0.34 |
| Hematocrit, % (range) | 38 (20–62) | 38 (27–42) | 37 (17–42) | 0.40 |
| WBC, n×109/L (range) | 14.1 (1.9–56.2) | 9.3 (6.9–32.2) | 10.5 (6.1–22.5) | 0.06 |
| Platelets, n×109/L (range) | 688 (49–1674) | 788 (153–1583) | 622 (121–1524) | 0.75 |
| LDH, U/L (range) | 805 (150–2505) | 727 (517–1886) | 643 (395–838) | 0.26 |
| Splenomegaly, n (%) | 13 (41.9) | 7 (54) | 1 (20) | 0.42 |
| Thrombotic events, n (%) | 13 (41.9) | 2 (15) | 2 (40) | 0.23 |
| Constitutional symptoms, n (%) | 8 (25.8) | 4 (31) | 3 (60) | 0.30 |
| Transfusion dependency, n (%) | 13 (41.9) | 0 (0) | 1 (20) | 0.02 |
| Stage, n (%) | 24 | |||
| Pre-fibrotic | 15 (48.3) | 9 (69) | 4 (80) | |
| Overt fibrotic | 16 (51.7) | 4 (31) | 1 (20) | |
| IPSS, n (%) | 0.09 | |||
| Low | 4 (12.9) | 7 (54) | 2 (40) | |
| Intermediate-1 | 12 (38.7) | 4 (31) | 2 (40) | |
| Intermediate-2 | 7 (22.6) | 0 (0) | 0 (0) | |
| High | 8 (25.8) | 2 (15) | 1 (20) | |
| DIPSS-plus, n (%) | 0.37 | |||
| Low | 4 (12.9) | 3 (23) | 1 (20) | |
| Intermediate-1 | 12 (38.7) | 7 (54) | 1 (20) | |
| Intermediate-2 | 11 (35.5) | 1 (8) | 1 (20) | |
| High | 4 (12.9) | 2 (15) | 2 (40) | |
| Karyotype, n (%) | 0.09 | |||
| Normal | 12 (100) | 7 (77.8) | 3 (60) | |
| Abnormal | 0 (0) | 2 (22.2) | 2 (40) | |
| NA | 19 | 4 | 0 |
PMF, primary myelofibrosis; WBC, white blood cell count; JAK2, Janus kinase 2; CALR, Calreticulin; MPL thrombopoietin receptor; WT, wild-type; MUT, mutated; LDH, lactate dehydrogenase; IPSS, International Prognostic Score System; DIPSS-plus, Dynamic International Prognostic Scoring System; NA, not available.
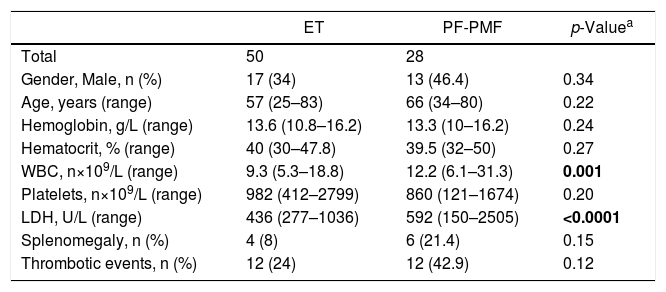
The comparison between ET and pre-fibrotic PMF (PF-PMF) in our cohort revealed that the latter presented a higher white blood cell count (p = 0.001) and LDH levels (p < 0.0001). It was also observed that PF-PMF patients had lower hemoglobin levels and a higher prevalence of splenomegaly, although these two findings were not statistically significant [Table 4]. There was no difference in the OS between ET and PF-PMF patients.
Clinical and laboratory features of ET and PF-PMF patients.
| ET | PF-PMF | p-Valuea | |
|---|---|---|---|
| Total | 50 | 28 | |
| Gender, Male, n (%) | 17 (34) | 13 (46.4) | 0.34 |
| Age, years (range) | 57 (25–83) | 66 (34–80) | 0.22 |
| Hemoglobin, g/L (range) | 13.6 (10.8–16.2) | 13.3 (10–16.2) | 0.24 |
| Hematocrit, % (range) | 40 (30–47.8) | 39.5 (32–50) | 0.27 |
| WBC, n×109/L (range) | 9.3 (5.3–18.8) | 12.2 (6.1–31.3) | 0.001 |
| Platelets, n×109/L (range) | 982 (412–2799) | 860 (121–1674) | 0.20 |
| LDH, U/L (range) | 436 (277–1036) | 592 (150–2505) | <0.0001 |
| Splenomegaly, n (%) | 4 (8) | 6 (21.4) | 0.15 |
| Thrombotic events, n (%) | 12 (24) | 12 (42.9) | 0.12 |
ET, essential thrombocythemia; PMF, primary myelofibrosis; PF, pre-fibrotic stage; WBC, white blood cell count; LDH, lactate dehydrogenase.
The current 2016 WHO classification for classical BCR-ABL1-negative MPNs is based on a multiparameter approach, including clinical features, bone marrow evaluation and genetic data. The latter is now considered one of the main criteria for diagnosis of these diseases and the frequency of the various phenotypic mutations changes according to the specific disease.
In our cohort, the JAK2V617 F mutation was found in almost 95% of PV patients, consistent with the literature. However, we detected one case with a JAK2 exon 12 mutation, previously reported,14,15 and one case with a JAK2 exon 9 mutation of uncertain pathogenicity, not previously described. Two cases had a JAK2 wild-type rare genotype with confirmed erythrocytosis, subnormal erythropoietin levels and bone marrow histopathology consistent with PV.13 In the approximately 3% of PV cases that do not present the JAK2V617 F mutation in exon 14, variant mutations spread across exons 12–15 have been described.15 By the use of Sanger sequencing, we did not observe any mutation in the JAK2 gene in two of our cases, although the WHO criteria for PV diagnosis was fulfilled. A further search for gene mutations associated with inherited polycythemias will be performed in order to better characterize those JAK2-negative PV cases.
Among the ET patients, JAK2V617F and CALR exon 9 mutations were found in 48% and 22% of the patients, respectively, consistent with the frequency described in the literature. However, the MPL exon 10 mutation was not found in any patients. Of note, two ET patients did not have the results of the MPL mutation search at the time the study data was analyzed. The absence of the MPL exon 10 mutation can also explain the higher frequency of triple-negative ET patients (22%) in our cohort, in comparison to what has been reported in the literature, although our numbers are similar to two other Brazilian studies that reported 27.6%16 and 31%17 of triple-negative ET patients. The presence of the JAK2V617F mutation in ET patients has been associated with a higher risk of thrombosis, hemoglobin levels and progression to PV or myelofibrosis.1,13,16,17 We found in our cohort that ET patients with this mutation had higher hemoglobin levels and a higher risk for disease than those other genotypes. However, the rate of thrombotic events in JAK2V617F ET patients was not higher than the one found in patients with other driver mutations.
Regarding the PMF patients, the frequency of JAK2V617F and CALR exon 9 mutations (60.8% and 23.5%, respectively) was also similar to that in previous reports. As in ET cases, the MPL exon 10 mutation was not found in the PMF patients. On the other hand, the frequency of triple-negative patients was slightly lower than the one reported in the literature. While the PMF patients with CALR exon 9 mutations have longer survival than the other genotypes, triple-negative PMF cases present with an aggressive disease and a high risk of transformation to AML.13 Rumi et al.12 found that CALR-mutated patients were significantly younger, had lower leukocyte counts, higher platelet counts, and a lower IPSS risk, while triple-negative patients were older, had lower hemoglobin levels, lower platelet counts, and a higher IPSS risk. They also found a median OS of 17.7 years in the CALR-mutant, 9.2 years in the JAK2-mutant, 9.1 years in the MPL-mutant, and 3.2 years in the triple-negative patients. In our cohort, we found the similar phenotypic features among the CALR-mutated and triple-negative patients, although we could not reach statistical significance, probably because of the low number of patients. The survival analysis among our PMF patients demonstrated that triple-negative patients had lower survival than those of other genotypes. However, the low number of patients and events may have compromised the global analysis.
The PF-PMF is considered the initial presentation of PMF and frequently displays thrombocytosis, implying a differential diagnosis with ET. The bone marrow biopsy represents the most important criteria for distinction between these two MPN phenotypes.3,13,18 Similarly to previous reports showing that PF-PMF was more frequently associated with elevated LDH, leukocytosis, anemia and splenomegaly than ET,19,20 the PF-PMF patients in our study presented more leukocytosis and elevated LDH than the ET patients. Such a confirmation is relevant because distinguishing diseases whose prognosis and treatment differ considerably is of utmost importance.
Although our report includes a limited number of patients, it is a single center study that represents a real-life characterization of a Brazilian MPN cohort that may guide not only research initiatives in this area of study, but also the clinical stratification and management of patients. Our results are consistent with the data from two other studies performed in southeastern Brazil.16,17 Further epidemiological studies of classical BCR-ABL1-negative MPNs cohorts in developing countries are encouraged in order to improve our knowledge on the clinical profile of these diseases, aiming at improving the local quality of care.
ConclusionsThis study grouped and classified Brazilian patients with classical BCR-ABL1-negative MPNs according to the new WHO diagnostic criteria for PV, ET and PMF. The frequency of somatic mutations according to the disease phenotype was consistent with previous reported data, despite some limitations. The clinical characteristics of this MPN cohort were observed and correlated with the mutation profile, prognosis and overall survival. We described two variant mutations in JAK2 (exons 9 and 12) among our PV cases. It was also possible to confirm the histological and clinical distinction between ET and PF-PMF. Further epidemiological studies of MPNs cohorts are required to improve the knowledge of the biology of these diseases in developing countries and to improve the quality of care.
Author contributionsM.A.P.S., R.D.O. and G.M.C. collected and interpreted the data and drafted the manuscript. J.A.M.N. performed the statistical analyses and drafted the manuscript. L.C.P. and L.L.F.P. gave the final approval of the version to be submitted. The manuscript was reviewed and approved by all authors.
Meeting of ethical standardsThis trial was approved by the institutional review board. The manuscript has not been published elsewhere and is not under submission at any other journal. All authors have agreed to the submission of this manuscript.
Conflicts of interestThe authors declare no conflicts of interest.
We thank all the members of the University Hospital of Ribeirao Preto Medical School and Center for Cell-Based Therapy laboratories for technical support in performing the mutational analysis.



