



Fanconi anemia (FA) is a rare autosomal recessive disease characterized by chromosomal instability and increased predisposition to malignancy. The diagnosis of FA requires clinical evaluation, confirmation of chromosomal fragility and/or analysis of genetic mutations. Therefore, this study aims to identify the clinical profile of patients with FA in the state of Pernambuco, Brazil.
MethodWe analyzed 100 individuals referred from the major hematology and bone marrow (BM) transplant centers in the state of Pernambuco, Brazil, between the years 2018 and 2022. The diagnosis of FA was performed using the mitomycin C chromosomal fragility test, clinical data and classical and molecular cytogenetic analyses.
ResultsWe enrolled a total of 16 patients with FA to comprise this study. Most of these individuals (87.5%) came from the Agreste and Sertão regions of Pernambuco. We observed a slight female prevalence of FA (1.3:1). The primary clinical and laboratory findings were café au lait spots (62.5%) and bone abnormalities (53%, mainly thumb deformities [40%]). We performed BM cytogenetic analysis for eight patients – seven showed no chromosomal abnormalities and one presented the karyotype 47,XY,+21 [15].
ConclusionsOur results are important to promote public health measures for the early diagnosis of FA, as well as to foster the engagement of a multidisciplinary group in the treatment of this disease.
Fanconi anemia (FA) is a rare autosomal recessive disease characterized by chromosomal instability. The classic clinical profile of this disease is highly variable: 90% of the patients have hematological abnormalities; 75% have physical and or skeletal deformities; 60% have delayed physical development; 25% have Fanconi-like facies; 40% have café au lait spots, and; 35% have thumb alterations.1 The etiopathogenesis of FA is related to a mutation in 22 genes responsible for DNA repair, as well as 13 genes responsible for ribosome biogenesis.2
The diagnosis of FA is based on clinical and molecular aspects, as well as chromosomal break hypersensitivity testing to alkylating agents such as diepoxybutane (DEB) and mitomycin C (MMC).3 Because of the consequences of bone marrow failure and the increased predisposition to malignancy, early identification of these individuals is essential for their appropriate clinical management.
The median survival of patients with FA in the USA was 19 years between 1981 and 1990. However, with early diagnosis and the introduction of new treatment modalities, survival from 1991 to 2006 increased to 30 years.4 There is no data regarding patient survival in Brazil, but Bonfin et al.5 reported an adapted protocol for hematopoietic stem cell transplantation (HSCT). This protocol was associated with an overall 5-year survival of 83%, good engraftment and a low incidence of acute graft-versus-host disease at day 100 post-transplantation.
Patients with FA have a 500-fold increased risk of developing myelodysplastic syndrome (MDS) and or acute myeloid leukemia (AML).6 About 80% of the patients evolve to MDS and or AML, the main indicators of this evolution being chromosomal alterations in bone marrow (BM), such as -7/del (7q), abnormalities in 3q and 11q and complex karyotypes.7
Approximately 1:181 Americans and 1:93 Israelis are affected by FA.8 Although FA can affect all races, it is rare in African descendants (1:476.000).9 In Brazil, two important groups in the states of São Paulo10,11 and Paraná,12 registered 148 and 52 patients with chromosomal breakage, respectively. Two other studies were conducted, with one describing the clinical profile of 17 patients with FA13 and another identifying mutations in the FANCA, FANCC and FANCG genes in 255 patients, 68 of whom were from the northeastern region of Brazil.14
The implementation of the MMC chromosomal fragility test has supplied the northeastern region of Brazil with an early and rapid diagnosis of FA cases, providing a targeted follow-up and increasing the patient's quality of life. However, there are no studies in loco with epidemiological data, forms of clinical presentation and cytogenetic profile of FA for this region. Therefore, conducting research on the FA and implementing the MMC chromosomal fragility test in patients of the Unified Health System (Sistema Único de Saúde [SUS]) in Pernambuco (PE) is relevant for Brazil, mainly for the northeastern region. Furthermore, it can provide specific regional information concerning the clinical, laboratory and cytogenetic characteristics of patients with FA.
Material and methodsPatients and sampleA prospective study was conducted between the years 2018 and 2022 to investigate the clinical profile of FA. One hundred individuals were investigated in this study and sixteen of them were diagnosed as Fanconi positive and followed in this study. admitted to the outpatient clinic at the Centro de OncoHematologia Pediátrica, Hospital Universitário Oswaldo Cruz for evaluation of clinical and family history, physical examination and laboratory tests. The individuals included were referred from the Fundação de Hematologia e Hemoterapia de Pernambuco, Instituto do Câncer do Agreste, Instituto de Medicina Integral Professor Fernando Figueira and Hospital Português – Centro de Transplante de Medula Óssea. Only patients with at least two of the following symptoms were included in the study: hypoplastic or aplastic BM, skeletal abnormalities, café au lait spots, and pancytopenia with an increase in the mean corpuscular volume or fetal hemoglobin.
The study protocol was approved by the local research ethics committee (approval number 73905817.1.1001.5192). The guardians of the children were informed about the tests to be performed and authorized participation, as well as the disclosure of findings, by signing the informed consent form. Data collection began after the approval of the study.
Mitomycin C chromosomal fragility testThe test protocol was adapted from Oostra et al.15 Each patient had 5mL of peripheral venous blood collected in a heparinized syringe. The lymphocyte culture was prepared according to the standard polyvinyl butyral (PVB) culture protocol with the addition of MMC at a concentration of 150 nM at the time of culture medium preparation. A total of 20 µL of 10-3 colcemid (Sigma) were added after a 70-hour culture in a CO2 incubator. The material was centrifuged and hypotonic shocked with potassium chloride for 15 minutes at 37 °C in a water bath after 72 hours and then fixed with methanol and acetic acid (3:1). The slides were made and stained with Giemsa (Merck) at 5% for 8 minutes. Twenty-five metaphases from each individual were evaluated and chromosomal variants, such as breaks, fragments, ring chromosomes, radial figures and rearrangements were analyzed.
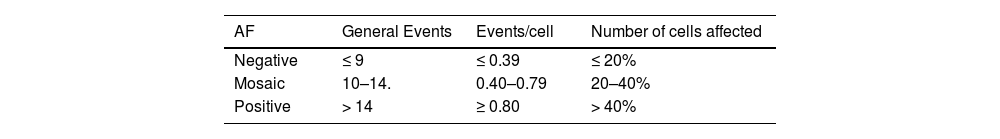
A score was assigned for each chromosomal variant in the following way: for each break and fragment, one point; for each ring and radial figure chromosome, two points, and; for each chromosome involved in the rearrangement, one point. The total points were added to produce a final score and the patients were classified as suggestive (> 40%) or not suggestive of FA (< 20%) based on this score, as shown in Table 1.
Bone marrow cytogenetic analysisThe cytogenetic analysis of the BM was performed using 24-hour cultures according to standard protocols. At least 20 metaphases in each case were analyzed by G-banding according to standard protocols and characterized in compliance with the International System of Human Cytogenetic Nomenclature (2020).16
The cytogenetic analysis could not be performed in all individuals because we did not have medical approval to perform the BM aspiration. Patients undergoing cytogenetic analysis are followed up every six months for medical evaluation.
Fluorescence in situ hybridizationFluorescent probes were used for the molecular cytogenetic technique, as per Liehr 17 and the manufacturer's instructions. Analyses were performed in metaphase and/or interphase cells using the centromeric probe of chromosome 21.
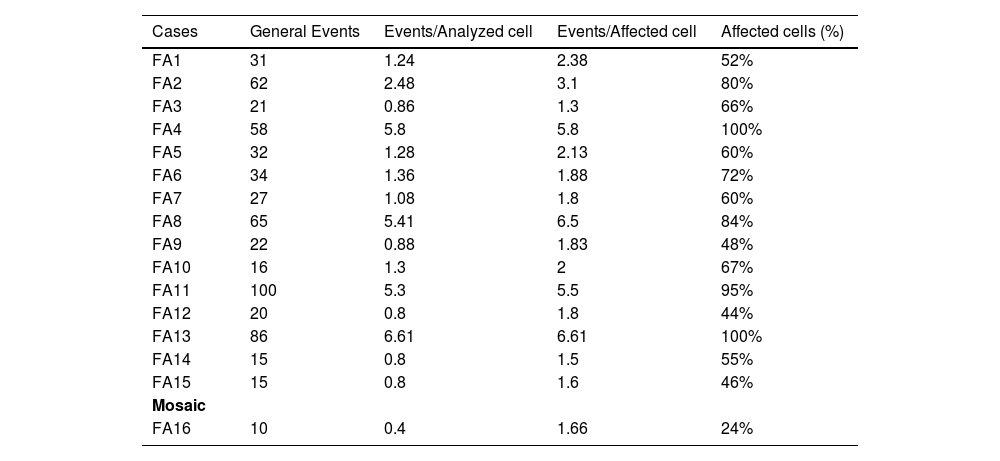
ResultsIn the present study, 100 individuals were referred for investigation of FA. Among those with a negative test, one patient was diagnosed with the thrombocytopenia absent radius (TAR) syndrome and another with the Pearson syndrome. A third patient, who had skeletal abnormalities of the thumb, renal agenesis, hemivertebrae and a positive MMC chromosomal fragility test, had a final diagnosis of vertebral defects, anal atresia, cardiac defects, trachea-esophageal fistula, renal anomalies and limb abnormalities (VACTERL)/FA association (FA3). The remaining 16 individuals were diagnosed as FA through the MMC chromosomal fragility test, as shown in Table 2. One patient (FA16) presented as a mosaic for FA.
Results of the fragility test by MMC.
| Cases | General Events | Events/Analyzed cell | Events/Affected cell | Affected cells (%) |
|---|---|---|---|---|
| FA1 | 31 | 1.24 | 2.38 | 52% |
| FA2 | 62 | 2.48 | 3.1 | 80% |
| FA3 | 21 | 0.86 | 1.3 | 66% |
| FA4 | 58 | 5.8 | 5.8 | 100% |
| FA5 | 32 | 1.28 | 2.13 | 60% |
| FA6 | 34 | 1.36 | 1.88 | 72% |
| FA7 | 27 | 1.08 | 1.8 | 60% |
| FA8 | 65 | 5.41 | 6.5 | 84% |
| FA9 | 22 | 0.88 | 1.83 | 48% |
| FA10 | 16 | 1.3 | 2 | 67% |
| FA11 | 100 | 5.3 | 5.5 | 95% |
| FA12 | 20 | 0.8 | 1.8 | 44% |
| FA13 | 86 | 6.61 | 6.61 | 100% |
| FA14 | 15 | 0.8 | 1.5 | 55% |
| FA15 | 15 | 0.8 | 1.6 | 46% |
| Mosaic | ||||
| FA16 | 10 | 0.4 | 1.66 | 24% |
The mean age at diagnosis was 8.5 years, with a slight female prevalence of FA (56.25%). Approximately 87.5% of patients were from the Agreste and Sertão regions of Pernambuco and 62.5% were born to consanguineous parents.
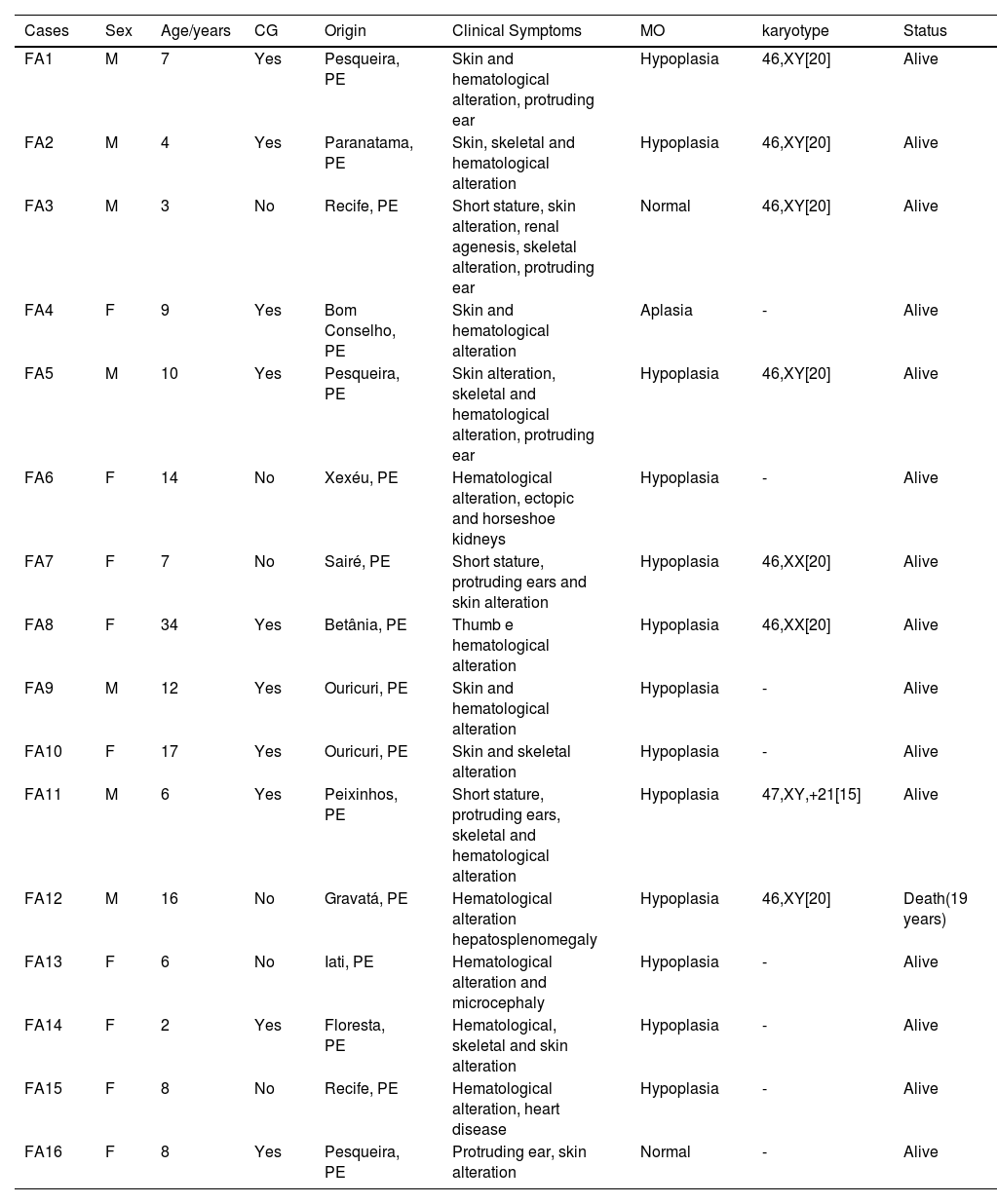
The clinical and laboratory profiles of FA (Table 3) showed that most patients had hematological alterations (80%), particularly medullary failure (87.5%). Hypoplasia of the BM was seen in 92.86% and aplasia in 7.14%. Café au lait spots occurred in 62.5% of the cases and 53% of the patients had skeletal changes, especially thumb hypoplasia (40%) (Figure 1). Fewer patients had short stature, protruding ears, microcephaly, heart disease and unilateral renal agenesis.
Clinical and demographic data of patients with FA.
| Cases | Sex | Age/years | CG | Origin | Clinical Symptoms | MO | karyotype | Status |
|---|---|---|---|---|---|---|---|---|
| FA1 | M | 7 | Yes | Pesqueira, PE | Skin and hematological alteration, protruding ear | Hypoplasia | 46,XY[20] | Alive |
| FA2 | M | 4 | Yes | Paranatama, PE | Skin, skeletal and hematological alteration | Hypoplasia | 46,XY[20] | Alive |
| FA3 | M | 3 | No | Recife, PE | Short stature, skin alteration, renal agenesis, skeletal alteration, protruding ear | Normal | 46,XY[20] | Alive |
| FA4 | F | 9 | Yes | Bom Conselho, PE | Skin and hematological alteration | Aplasia | - | Alive |
| FA5 | M | 10 | Yes | Pesqueira, PE | Skin alteration, skeletal and hematological alteration, protruding ear | Hypoplasia | 46,XY[20] | Alive |
| FA6 | F | 14 | No | Xexéu, PE | Hematological alteration, ectopic and horseshoe kidneys | Hypoplasia | - | Alive |
| FA7 | F | 7 | No | Sairé, PE | Short stature, protruding ears and skin alteration | Hypoplasia | 46,XX[20] | Alive |
| FA8 | F | 34 | Yes | Betânia, PE | Thumb e hematological alteration | Hypoplasia | 46,XX[20] | Alive |
| FA9 | M | 12 | Yes | Ouricuri, PE | Skin and hematological alteration | Hypoplasia | - | Alive |
| FA10 | F | 17 | Yes | Ouricuri, PE | Skin and skeletal alteration | Hypoplasia | - | Alive |
| FA11 | M | 6 | Yes | Peixinhos, PE | Short stature, protruding ears, skeletal and hematological alteration | Hypoplasia | 47,XY,+21[15] | Alive |
| FA12 | M | 16 | No | Gravatá, PE | Hematological alteration hepatosplenomegaly | Hypoplasia | 46,XY[20] | Death(19 years) |
| FA13 | F | 6 | No | Iati, PE | Hematological alteration and microcephaly | Hypoplasia | - | Alive |
| FA14 | F | 2 | Yes | Floresta, PE | Hematological, skeletal and skin alteration | Hypoplasia | - | Alive |
| FA15 | F | 8 | No | Recife, PE | Hematological alteration, heart disease | Hypoplasia | - | Alive |
| FA16 | F | 8 | Yes | Pesqueira, PE | Protruding ear, skin alteration | Normal | - | Alive |
CG: Consanguineous marriage; MO: Bone marrow; M: Male; F: Female.
 Hyperpigmentation on the face; b) Hypoplasia of the thumb; c) skeletal malformation in the feet.")
Cytogenetic studies of BM were performed on eight of 16 patients. Seven individuals had normal karyotype and one (FA11) had karyotype 47,XY,+21[15]/46,XY[5] (Figure 2). This patient had a phytohemagglutinin (PHA)-stimulated peripheral blood lymphocyte culture with normal karyotype (46.XY[5]). The main criteria described for the indication for HSCT include unfavorable cytogenetic changes, such as gain or loss of 1q, 3q, and 11q, -7, del7q and +8 and worsening of pancytopenia, with the need for transfusions or antibiotic. Thus, at the moment, the remaining patients still did not present criteria for HSCT indication and are in outpatient follow-up.
 Karyotype with G Banding: 47,XY,+ 21 b) FISH showing +21.")
One patient died of sepsis (FA12) during the present study and two others (FA2 and FA7) are alive after the HSCT. The remaining individuals were treated with Danazol, but to date, there has been no complete recovery from the BM hypoplasia. Furthermore, these patients had side effects, such as virilization, hypertrichosis and incomplete response.
DiscussionThe FA is a rare and poorly studied disease in Brazil and studies involving a clinical approach and cytogenetic and molecular profiles of this disease in the northeastern region are even scarcer. Most of the patients analyzed in previous studies were from the southern state Paraná and the southeastern state São Paulo.
Sixteen individuals were diagnosed with FA in the state of Pernambuco over four years, meaning 4.5 cases/year, with a mean age at diagnosis of 8.5 years. From 1995 to 2012, Pilonetto et al.14 studied 68 cases of FA in the northeastern region of Brazil and observed a rate of 3.4 cases/year. This underreporting observed may be explained by the difficulty in sending samples for MMC chromosomal fragility testing to the southern region of Brazil because this test was not available in the northeastern region.
A difference in the age range at diagnosis was observed when comparing our results with the findings of Shimamura and Alter.18 These authors described that the mean age at diagnosis of FA was 6.5 years, whereas our study showed a mean age of 8 years (minimum of 2 years and maximum of 34 years). This demonstrates a deficit in the early diagnosis of the disease. However, when compared to a previous study in which the mean age was 13 years, it was possible to observe a 5-year decrease in the diagnosis with the start of the MMC chromosomal fragility test in Pernambuco, which provided the patient with better follow-up, treatment and survival time (personal communication).
There is no consensus on the prevalence of FA between the sexes. Some authors stress that there is an equal distribution,19,20 whereas others found a higher male prevalence.1,21,22 The present study demonstrated a discrete female prevalence of the FA (1.3:1) in Pernambuco, Brazil.
Among the 16 patients with a confirmed diagnosis of FA, 62.5% were born of consanguineous marriages. This demonstrates that consanguinity is a relevant factor for the predisposition to the disease, as well as increasing its prevalence in the overall population.20 The high level of consanguinity observed in our state, Pernambuco, is probably related to the region of origin of these patients, as 87.5% of them come from the Agreste and Sertão regions.
The clinical profile of FA identified in the current research was the same as that reported by Santos.23 According to Santos, approximately 75% of patients with FA present congenital defects, such as pigmentation and café au lait spots on the skin, described in more than 50% of the patients, short stature in about 50% and skeletal anomalies in 30%. Zen et al.13 also reported thumb alterations (60%) and café au lait spots (43%) as the primary alterations, in addition to renal agenesis. This study reported all these clinical presentations for one patient.
Acquired chromosomal changes are frequent findings in BM karyotypes of patients with FA undergoing clonal evolution to MDS or AML. Borges et al.24 reported four patients with FA undergoing transformation to MDS (three cases) and AML (one case) who showed chromosomal changes in the BM, one being recurrent and the others, new translocations. Acquired chromosomal changes are the main indication for BM transplantation and the main changes described are losses and gains in the 1q, 3q, and 11q regions, as well as in -7 and +8.25 However, in this study, only one patient (FA11) showed acquired trisomy 21, which may indicate clonal evolution. Unlike de novo acute leukemias, alterations involving chromosome 21 have been poorly described for FA.
Ayas and Walter26 described a case of FA with a complex karyotype in which dup(21)(q22)x2,+add(21)(q22) was observed. In this duplicated region, the AML1 gene (RUNX1) is present, which is correlated with myeloid neoplasms, suggesting an important role of this gene in characterizing disease progression. Another case of FA with monosomy 21, (21q22.1-q22.1) was reported by Byrd et al.,27 resulting in monosomy of the RUNX1 gene that is directly related to thrombocytopenia and defective platelet functionality.28 Additionally, four other cases of chromosomal alteration involving chromosome 21 in patients with FA have been described by Quentin et al.,29 two of which presented complex karyotypes 46,XX,der(1)t(1;21)(p36;q22).ish(RUNX1+,PRDM16+),der(2)t(2;3)(q37q21),der(16)t(1;16)(q21;q12)[20] and 46,XY,add(21)(q21)[1] /46,XY,der(7)t(7;21)(q32;q22)[3]; one, the deletion 46,XX,del(21)(q22.12), and; one, the duplication 46,XX, dup(21)(q22.12-q22.3). All four patients progressed to MDS or AML, confirming the important role of chromosome 21, particularly the RUNX1 gene (21q.22), in the etiopathogenesis of progression to acute leukemia.
In the present study, patient FA11 with trisomy 21 did not have Down syndrome (stimulated peripheral blood karyotype did not detect +21). The occurrence of acquired trisomy 21 led to the overexpression of the RUNX1 oncogene in this patient and possibly a stage of clonal evolution to MDS and/or AML. The clinical phenotype of patients with FA is highly heterogeneous, which makes diagnosis difficult because they present symptoms common to several other pathologies, such as the VATER, or VACTERL, Nejimegen, Seckel, thrombocytopenia, TAR, Pearson, Bloom and ataxia-telangiectasia syndromes and Diamond-Blackfan anemia.30 Syndromes, such as the Nejimegen, Seckele and Bloom, may have chromosomal breaks by DEB and MMC in smaller numbers than in FA cases and may be confused with heterozygous patients. For these cases, the search for a specific genetic mutation is mandatory for an accurate diagnosis.1 Therefore, patients with chromosomal fragility tests non-compatible with FA, but who have low fragility index and signs suggestive of other pathologies, should always undergo differential diagnosis with another chromosomal fragility disease.
ConclusionOur results have relevance for the SUS and help in the early recognition of FA. Therefore, we hope that this study encourages public health measures aimed at achieving early diagnosis and improving the care and treatment of patients and their families. We highlight the importance of monitoring the BM by cytogenetic studies in these patients, aiming at the early identification of possible signs of worsening or disease progression to MDS or AML. The HSCT is an important therapeutic modality for patients with these disease progressions.
The authors thank the patients, families and doctors for the data. We are also grateful for the partial support supplied by Coordenação de Aperfeiçoamento Pessoal de Nível Superior (CAPES) for this study, Finance Code 001.


