



Chimeric Antigen Receptor T (CAR-T) cells are certainly an important therapy for patients with relapsed and/or refractory hematologic malignancies. Currently, there are five CAR-T cell products approved by the FDA but several research groups and/or biopharmaceutical companies are encouraged to develop new products based on CAR cells using T or other cell types. Production of CAR cells requires intensive work from the basic, pre-clinical to translational levels, aiming to overcome technical difficulties and failure in the production. At least five key common steps are needed for the manipulation of T-lymphocytes (or other cells), such as: cell type selection, activation, gene delivery, cell expansion and final product formulation. However, reproducible manufacturing of high-quality clinical-grade CAR cell products is still required to apply this technology to a greater number of patients. This chapter will discuss the present and future development of new CAR designs that are safer and more effective to improve this therapy, achieving more selective killing of malignant cells and less toxicity to be applied in the clinical setting.
Currently, there are five CAR-T cell products approved by the FDA and produced by big pharmaceutics’ companies using automated or semi-automated systems, allowing the manufacturing of CAR-T cells at a large-scale level. Nevertheless, other distinct protocols using similar or different technologies for the generation of autologous or allogeneic CAR-T cells or CAR-NK cells are currently under development. Manufacturing of these cells require an intensive work from the basic, pre-clinical to translational levels, aiming to overcome technical difficulties and failure in the production.1 Characteristics of the final product, including the number of obtained modified cells, and its degree of activation and functionality, may also vary accordingly to the distinct outlines and patients’ intrinsic features.
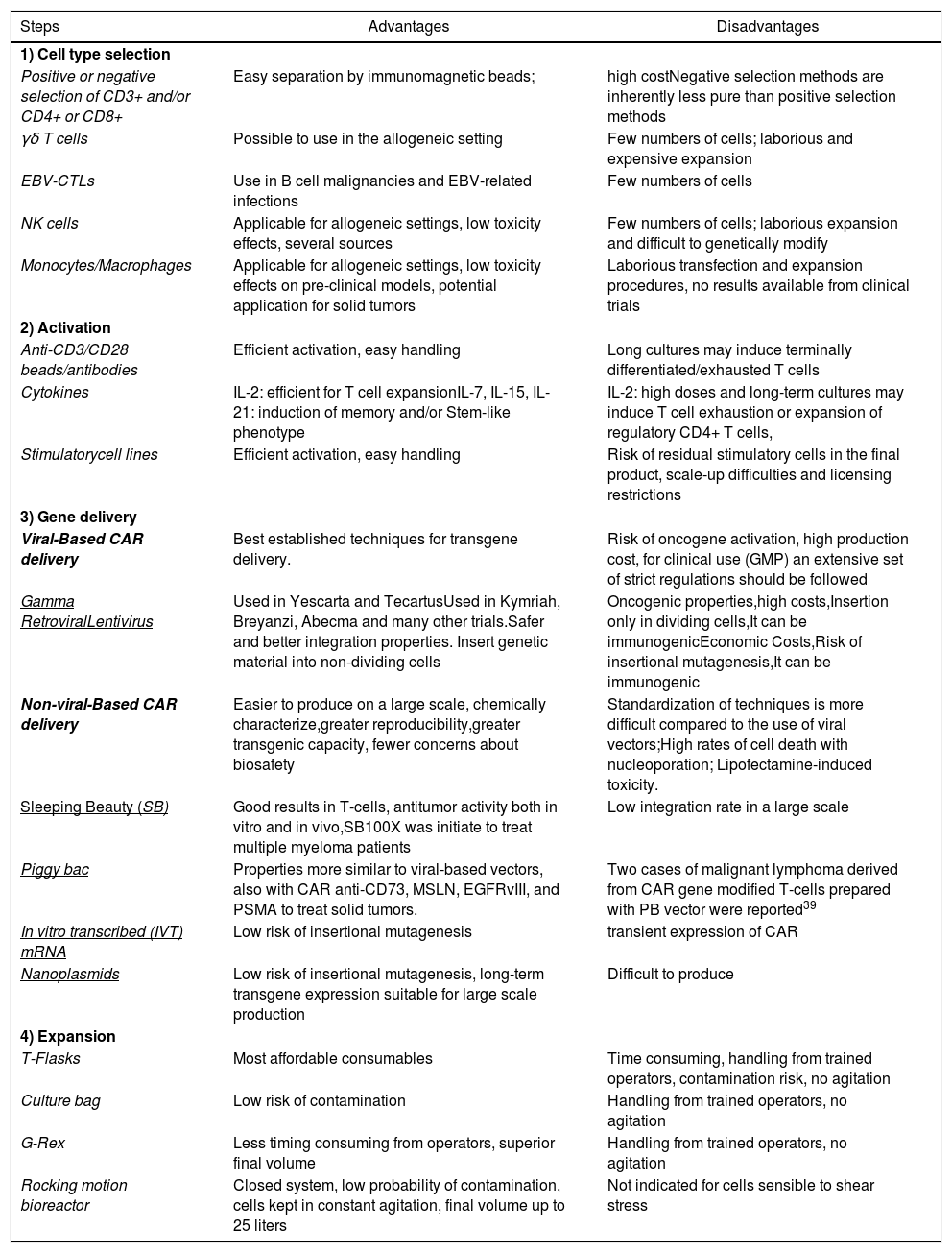
In general, the production of autologous CAR-T cells requires at least five key common steps for the manipulation of T-lymphocytes, such as: cell type selection, activation, gene delivery, cell expansion and final product formulation (Figure 1; Table 1). Subsequently to the manufacture, a quality control testing is required prior the infusion of CAR-T cells into the patients.

Advantages and disadvantages of the technologies for CAR cell production.
| Steps | Advantages | Disadvantages |
|---|---|---|
| 1) Cell type selection | ||
| Positive or negative selection of CD3+ and/or CD4+ or CD8+ | Easy separation by immunomagnetic beads; | high costNegative selection methods are inherently less pure than positive selection methods |
| γδ T cells | Possible to use in the allogeneic setting | Few numbers of cells; laborious and expensive expansion |
| EBV-CTLs | Use in B cell malignancies and EBV-related infections | Few numbers of cells |
| NK cells | Applicable for allogeneic settings, low toxicity effects, several sources | Few numbers of cells; laborious expansion and difficult to genetically modify |
| Monocytes/Macrophages | Applicable for allogeneic settings, low toxicity effects on pre-clinical models, potential application for solid tumors | Laborious transfection and expansion procedures, no results available from clinical trials |
| 2) Activation | ||
| Anti-CD3/CD28 beads/antibodies | Efficient activation, easy handling | Long cultures may induce terminally differentiated/exhausted T cells |
| Cytokines | IL-2: efficient for T cell expansionIL-7, IL-15, IL-21: induction of memory and/or Stem-like phenotype | IL-2: high doses and long-term cultures may induce T cell exhaustion or expansion of regulatory CD4+ T cells, |
| Stimulatorycell lines | Efficient activation, easy handling | Risk of residual stimulatory cells in the final product, scale-up difficulties and licensing restrictions |
| 3) Gene delivery | ||
| Viral-Based CAR delivery | Best established techniques for transgene delivery. | Risk of oncogene activation, high production cost, for clinical use (GMP) an extensive set of strict regulations should be followed |
| Gamma RetroviralLentivirus | Used in Yescarta and TecartusUsed in Kymriah, Breyanzi, Abecma and many other trials.Safer and better integration properties. Insert genetic material into non-dividing cells | Oncogenic properties,high costs,Insertion only in dividing cells,It can be immunogenicEconomic Costs,Risk of insertional mutagenesis,It can be immunogenic |
| Non-viral-Based CAR delivery | Easier to produce on a large scale, chemically characterize,greater reproducibility,greater transgenic capacity, fewer concerns about biosafety | Standardization of techniques is more difficult compared to the use of viral vectors;High rates of cell death with nucleoporation; Lipofectamine-induced toxicity. |
| Sleeping Beauty (SB) | Good results in T-cells, antitumor activity both in vitro and in vivo,SB100X was initiate to treat multiple myeloma patients | Low integration rate in a large scale |
| Piggy bac | Properties more similar to viral-based vectors, also with CAR anti-CD73, MSLN, EGFRvIII, and PSMA to treat solid tumors. | Two cases of malignant lymphoma derived from CAR gene modified T-cells prepared with PB vector were reported39 |
| In vitro transcribed (IVT) mRNA | Low risk of insertional mutagenesis | transient expression of CAR |
| Nanoplasmids | Low risk of insertional mutagenesis, long-term transgene expression suitable for large scale production | Difficult to produce |
| 4) Expansion | ||
| T-Flasks | Most affordable consumables | Time consuming, handling from trained operators, contamination risk, no agitation |
| Culture bag | Low risk of contamination | Handling from trained operators, no agitation |
| G-Rex | Less timing consuming from operators, superior final volume | Handling from trained operators, no agitation |
| Rocking motion bioreactor | Closed system, low probability of contamination, cells kept in constant agitation, final volume up to 25 liters | Not indicated for cells sensible to shear stress |
In this chapter we summarize the key steps for autologous CAR (-T or others) cells production including some novel technologies that are under development or in phase I/II clinical trials.
Key steps for CAR cell manufacturing- 1)
Cell type selection
The first step in CAR-T cell production is obtaining donor/patient T lymphocytes from peripheral blood mononuclear cells (PBMCs) using whole blood samples or a leukapheresis product. Subsequently, researchers might isolate CD3 positive T cells or other specific subset of these cells. The most common approach to enrich for target T cells is by immunomagnetic separation (IMS), using magnetic microbeads conjugated to antibodies specific for markers of target (positive) or non-target (negative) cells in order to separate a specific population.2 Usually, T cells are positively selected with CD3+ marker but CD4+ and CD8+ selection can also be applied to specific isolation of helper or cytotoxic phenotypes, especially when specific CD4:CD8 ratios are intended.3
Other T cell subsets are of special interest due to some particularities that might improve CAR-T cell function. Gamma-delta T cells (γδ) have being explored as a specific group that exhibit a natural anti-tumor reactivity and do not require antigen presentation via MHC complex for recognition and activation, increasing the scope of CAR-T cell application in the allogeneic context.4 Besides, the expansion of these cells in culture has been proven feasible with the application of IL-2 and bisphosphonates.4,5
Antigen-specific cytotoxic T cells (CTLs) such as Epstein-Barr Virus (EBV)-CTLs, whose efficacy on the immunotherapy setting has been widely demonstrated, are also being explored as a source for CAR-T cell production. The co-cultivation of PBMCs with irradiated EBV B-lymphoblastoid cell lines (LCLs) enriches for EBV-CTLs in culture, allowing their expansion and further engineering for applications in B cell malignancies and EBV-related infections6 (Table 1).
Alternatively, other cell types are currently under evaluation in pre-clinical or phase 1-2 trials, such as macrophages and NK cells. Klichinsky et al have reported the insertion of anti-HER2 CARs in both THP-1 macrophage cell lines and primary human macrophages (CAR-M).7 Anti-HER2 CAR-M were efficient for ovarian tumor cells phagocytosis and elimination in vitro and in pre-clinical xenograft models of SKOV3 tumor cells. Additionally, Liu and collaborators have recently published the first phase 1-2 trial using CAR-NK cells derived from cord blood cells.8 Eleven patients with relapsed chronic lymphocytic leukemia or non-Hodgkin's lymphoma were treated with anti-CD19-IL-15 CAR-NK cells and data indicated no adverse effects or toxicity for any of the individuals. Further, around 70% of the patients responded to treatment and CAR-NK cells were found at patients’ circulation for around 12 months.
- 2)
Activation
Moloney Leukemia Virus (NLV) -based Retroviral vectors require target cells to be cycling to allow gene transfer while HIV-based lentiviral vectors benefit from T cell proliferation to achieve high efficiency T cell transduction.9 Complete activation of T cells require signal 1 (CD3) and 2 (CD28) engagement,10 which can be achieved with the employment of anti-CD3 and anti-CD28 monoclonal for in vitro T cell activation for gene modification. A crucial contribution came later when beads were developed as a support for anti-CD3/CD28 antibodies immobilization,11 setting the basis for most of the in vitro T cell transduction protocols,12 with cytokine supplementation (usually IL-2) largely applied to these protocols (Table 1).
Stimulating and culturing T cells for long period of time can lead to terminal differentiation. To overcome this limitation, and in order to optimize the phenotype of therapeutic T cells, some groups started reducing the culturing time13,14 or adding cytokines that can help retaining a more memory and stem phenotype. Such cytokines include, but are not restricted to, IL-7, IL-1515 and IL-21.16 Such stem-phenotype-based protocols can potentially result in shorter expansion protocols and the infusion of a lower number of cells to achieve the same biological effect, since these cells expand and differentiate in vivo, sustaining the antitumor response.15
This cytokine signaling can be provided by soluble molecules added to the culture or by engineered cell lines that display several of these signals in the membrane, such IL-15, several co-stimulating molecules like CD80 and even CAR-target molecules such as CD19.17 The stimulating artificial antigen presenting cells (aAPC) can represent a valuable tool for the generation of CAR-T cells. A variation of this strategy consists in using LCLs as stimulating cells with the possibility of generating CAR-T cells that target the tumor antigen and also viral antigens. Some examples of these protocols have been already reported in the preclinical18 or clinical setting.19
Other than cytokines, several groups are characterizing different molecules that can be used to retain better phenotypes for CAR-T cells to perform as potent antitumor agents once infused in patients. These molecules include some AKT20 or PI3K21 inhibitors amongst other candidates.
- 3)
Gene Delivery
The success of CAR-T cell therapy depends on the selection of an appropriate vector that will transport the CAR construct into the cells (Table 1). The two most commonly used options are virus-based vectors (retroviruses or lentiviruses) or non-viral vectors, which are predominantly transposon vectors (Figure 2A). Currently, there are five launched CAR-T products and all of them use viral vectors to introduce CAR into T cells (Figure 2B).
 Vector systems used in CAR-T cell therapies. B) CAR-T products currently on the market. Data available in the Integrity database (Clarivate Analytics).")
Two of the CAR-T products currently available in the market are based on retroviral vectors (Yescarta and Tecartus). Vector-based on murine leukemia virus (MLV) is the most commonly used gamma retroviral vector22 and it has been successfully applied in other therapies such as T-cell immunotherapy for combined immunodeficiency- (SCID) X1 disease.23 Although, immunodeficiency disease has been successfully treated, in some patients, leukemia has been caused by the integration site of retroviral vector.23,24 Gamma retroviral vectors tend to integrate close to promoter regions, which may be the cause of their oncogenic properties. On the other hand, lentiviral vectors, derived from the retroviral family, Lentiviridae, showed safer and better integration properties than their gamma retroviral counterparts.25,26 Lentiviral vectors are able to insert genetic material into non-dividing cells,27 whereas gamma retroviral vectors transduce only dividing cells.28 The advantages of the lentiviral system are reflected in the large number of CAR-T products under development using this system and three of the CAR-T products available on the market use lentiviral vectors (Kymriah, Breyanzi and Abecma). (Table 1).
Since the beginning of CAR-T cell therapy, viral vectors have demonstrated high transduction efficiency. However, this type of vector presents i) a risk of insertional mutagenesis,29 ii) it can be immunogenic and iii) its manufacture is extremely expensive.30
Non-viral-based CAR deliveryAn alternative to viral vectors is the transposon system. A transposon is a sequence of DNA with the ability to change position within a genome via transposase excision and insertion.3 A variety of transposon-based systems have been reported for the production of CAR-T cells. The sleeping beauty (SB) transposon system showed good results in T-cells modified with anti-CD19 CAR, presenting antitumor activity both in vitro and in vivo.18 The main obstacle to using the SB transposon on a large scale is its low integration rate. New systems such as the SB1 and SB100X have much higher transposition rates than the native SB transposon.31,32 SB was the first non-viral vector to be used in clinical trials to generate CD19-specific CAR-T cells for treatment of NHL (Non-Hodgkin lymphoma) and ALL (Acute lymphocytic leukemia) (NCT00968760, NCT01497184) and recently, a new clinical trial with SLAMF7 CAR-T cells prepared with SB100X was initiate to treat multiple myeloma patients.33 Although the integration profile of SB can be considered biologically safe, other transposons such as PiggyBac (PB) transposon demonstrated properties more similar to viral-based vectors.31 PB were successfully used in the generation of CAR T-cells against CD19 to treat hematological malignancies34 and also with CAR anti-CD73,35 MSLN,36 EGFRvIII,37 and PSMA38 to treat solid tumors (Table 1).
Importantly, two cases of malignant lymphoma derived from CAR gene modified T-cells were described in patients that received CAR-T cells anti-CD19 prepared with PB vector,39 emphasizing the need for regular monitoring of new gene transfer methods used in clinical immunotherapy.
The risk of insertional mutagenesis has driven the development of other innovative gene delivery methodologies such as nanocarrier that delivers in vitro-transcribed (IVT) CAR and nanoplasmid. In vitro transcribed (IVT) mRNA has emerged as a new class of disruptive drugs that can be used to delivery CAR into T-cells. Several ongoing clinical trials are testing the efficiency and safety of engineered CAR mRNA T-cells to treat cancer patients (NCT01355965, NCT01897415, NCT02277522 and NCT02624258), and data suggest transient expression of CAR after infusion of cells is already enough to trigger antitumor responses.40
Nanoplasmids are emerging as a safer alternative to integrating vectors. Plasmid vectors based on S/MAR (scaffold and matrix attachment region) allow plasmid retention in the host cell nucleus, through interactions with nuclear matrix proteins ensuring proper plasmid segregation at mitosis.41 Bozza et al. 2021 demonstrates that this type of vector can be used efficiently to manipulate human T lymphocytes, support long-term transgene expression, can be scaled up, and is suitable for clinical use.42
Innovations to optimize vectors that can be used in CAR-T cell therapy will certainly continue to emerge.
- 4)
Expansion
The conventional protocols for T-cells activation/expansion ex-vivo include a combination of the three steps for optimal quality of the final product: activation strategy, gene delivery and expansion. The phenotypic and functional profiles of CAR-T cells may vary accordingly to the outline used for expansion as well as patient intrinsic features. Some studies have suggested advantages in the development of CAR-T cells presenting phenotypes such as stem cell memory43 and central memory,44 thus, the processes of expansion of CAR-T cells is critical for the quality of the final product. In fact, the choice of culture outline for expansion represents one of the main factors for T-cell expansion. As reviewed by Vormittag and collaborators,3 the most used culture systems reported in clinical trials may be categorized into three main types: 1) plates or T- flasks, present in 22% of the studies; 2) Static culture bags, found in 35% of the reports; and 3) Rocking motion bioreactor, reported by 43% of the clinical trials. Advantages and disadvantages of each of these culture conditions are listed in Table 1.
Considering the variation in workflows for the obtaining of a satisfactory number of cells for infusion, there is an urgent need of standardization of clinical protocols.
- 5)
Final formulation/product
The final formulation of products involves critical validation steps prior to cryopreservation. These tests aim to check safety, purity and potency, including T cells viability and CAR expression in the cell surface. The cryopreservation formula might contain 5 to 10% clinical grade DMSO, along with an electrolytic solution, human serum albumin and colloid solution in various combinations. Also, freezing rate should be between one and two Celsius degrees per minute, at least until -40°C, when the product could then be transferred to a liquid nitrogen tank. The CAR-T products should also be released under sterility and purity tests in order to ensure the product's safety. The standard protocols consist in testing for bacterial, fungal and mycoplasma contamination as well as for endotoxins.
Novel approaches for CAR-T cell productsModulation of CAR-T functionsA series of limitations have been reported in the use of CAR-T cells therapy. One main concern is the uncontrolled magnitude of CAR-T cells’ activation and expansion after infusion.45,46 In addition, CD19-specific CAR-T cells responding patients also shown B-cell aplasia and, consequently, hypogammaglobulinemia, a complication that usually requires periodic intravenous immunoglobulin administration. To overcome these complications, recent studies have described strategies to turn-on and off CAR-T cells.
Among these approaches, some groups engineered regulatory switches to inhibit or promote the infused cells activation. Fedorov and collaborators47 developed an inhibitory-CAR (iCAR) system by combining the antigen recognition domain with the intracellular inhibitory signaling via the receptors CTLA-4 and PD-1, two molecules involved in T-cell suppression. This strategy leads to a limited T lymphocyte capability, reducing T cell proliferation, cytotoxicity and cytokine production. Another series of studies applied the use of small-molecules drugs able to control the magnitude of CAR-T cells activation.48-50 Recently, Jan and colleagues reported the use of lenalidomide in an inducible on- vs off-switch system for CAR-T cells activation and degradation, respectively. For CAR-T receptor degradation, an IKZF3 zinc finger degron tag was inserted in the intracellular domains from 4-1BB and CD3z, which became accessible for lenalidomide binding. This will lead to ubiquitin ligase recruitment, followed by a polyubiquitination and subsequently transient CAR degradation. Conversely, in the ON-switch outline, authors constructed a two-component CAR: one subunit of the CAR containing the CD28 transmembrane and the intracellular domains composed by the lenalidomide-dependent zinc finger from IKZF3; and the second subunit composed by a CD28 domain, a mutated CRBN and a CD3-zeta signaling domain. In this settings, lenalidomide-dependable dimerization induced about fivefold increasing of CAR-T cell activation.
Bispecific/Trispecific CAR-T cellsA series of recent studies have shown the emergence of neoplastic B cells lacking CD19 expression in an important proportion of anti-CD19 CAR-T cells treated patients, suggestive of a scaping mechanism.51,52 Recent strategies reported CAR-T cells carrying bispecific or trispecific TCR structures, able to simultaneously target distinct tumor-antigens. The first phase-1 clinical trial was reported recently by Shah and collaborators53 in the use of bispecific anti-CD19/CD20 CAR-T cells to treat patients presenting B cell non-Hodgkin lymphoma or chronic lymphocytic leukemia. Using CliniMACS Prodigy system (Miltenyi), non-cryopreserved bispecific anti-CD19/CD20 CAR-T cells were tested in 22 patients. Results indicate a high response rate (82%) and a low cytotoxicity rate (5%) of cytokine release syndrome.
Another strategy published recently reported promising in vitro and pre-clinical results by using trispecific CAR-T cells able to target CD19, CD20 and CD22 antigens. Fousek et al.54 reported the construction of a trispecific anti-CD19/CD20/CD22 CAR using a single transgene expressing the three molecules with 4–1BB and CD3ζ endodomains. Results obtained from in vitro and in vivo assays showed that trispecific anti-CD19/CD20/CD22 CAR-T cells presented superior killing activity and tumor control when compared to single anti-CD19 CAR-T cells, even in tumor cell lines lacking CD19 molecule. Accordingly, Schneider and colleagues55 reported a newly engineered trispecific anti-CD19/CD20/CD22 CAR-T using lentiviral vectors by testing multiple intracellular T cell signaling motifs. Trispecific anti-CD19/CD20/CD22 CAR-T cells were able to efficiently eliminate and produce cytokines when incubated to a variety of tumor cell lines expressing distinct levels of CD19, CD20 and CD22. In pre-clinical assays, engineered Raji cells expressing heterogeneous levels of CD19, CD20 and CD22 molecules were completely eliminated in NSG-mice in around fourteen days post-infusion.
Automated production and CAR deliveryThe routine of an “open” system for CAR-T cell production is still highly laborious and expensive. The emergence of fully automated closed systems was of great contribution to overcome these challenges.
The CliniMACS® Prodigy system, released in 2016 by Miltenyi Biotec, is one good example of how this type of system can be feasible applied for cell therapy protocols facilities. The main technological advantage offered by this system is the fully closed automation of at least four steps of CAR-T cell production (cell separation, activation, transduction and expansion) in a single module machine in around 10 days of production.56
Pre-clinical studies using Prodigy machine successfully show that cells transduced and expanded in this system could eliminate tumoral cells in vitro and in vivo. When exposed to CD19+ tumoral cells, the CAR+ cells produced high levels of IFN-gamma, TNF-alpha and Granzyme-B, proving their anti-tumoral effect.57,58
Two recent reports from a clinical trial were published for adult or pediatric refractory CD19+ leukemia and lymphoma (NCT03144583) using autologous CAR-T cell product transduced with a lentiviral vector via Prodigy system.59,60 In 2020, they treated 34 patients with ALL, CLL and NHL (adult and pediatric) with a maximum expansion of 2.16 × 106 cells with 48.2% of CAR+ cells. Cell viability after production ranged from 90 to 100% and all final products were completely free of biological and chemical contaminants. In the second report, in 2021, a total of 58 patients were treated in the same standardized workflow. Results showed around 71% of the patients presenting complete response after 100 days of infusion.
Another option for automated T cell expansion is the Cytiva's Xuri Cell Expansion System W25. This system is composed of a temperature-controlled rocking tray aiming to provide gentle mixing and aeration to the cell culture bag. The equipment is assisted by software for the adjustment of several cell culture parameters (e.g., temperature, rocking angle and gas concentration). The latest version accepts a variety of cell culture applications (T, NK and other adherent cells) and expands in a volume up to 25 liters.61
Concluding remarksCAR-T cells are certainly an important therapy for patients with relapsed and/or refractory hematologic malignancies. In addition, due to the market launching of five CAR-T cell products positioned as reliable options for the treatment of cellular malignancies, several research groups and/or biopharmaceutical companies were encouraged to develop new products based on CAR-T cells. However, reproducible manufacturing of high-quality clinical-grade CAR-T cell products is still required to apply this technology to a greater number of patients. Isolation, genetic modification and expansion of T cells are key steps to the successful manufacturing of CAR-T cells. The development of new CAR designs that are safer and more effective is needed to improve this therapy, achieving more selective killing of malignant cells.
RNR, TGMO and VGR are supported by CNPq# 442676/2020-4, VPC is supported by FAPESP# 2019/25309-0, FAPESP# 2013/08135-2, CNPq# 442484/2020-8. MHB is supported by grants of CNPq and FAPERJ.



