


The POEMS syndrome is a rare disorder characterised by polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy and skin manifestations. Castleman disease can be present in POEMS patients. Castleman disease was named after Benjamin Castleman, who first described the specific histological findings of angiofollicular lymph node hyperplasia in a lymph node in 1954.1 It is a group of heterogenous lymphoproliferative disorders which can be divided into three histological subtypes, namely hyaline vascular, plasma cell and mixed type. The concept of the Castleman disease variant of the POEMS syndrome was first described by Dispenzieri in 2014.2 About 11–30% of the patients who were diagnosed with the POEMS syndrome have concomitant Castleman disease.3 This case report aims to highlight the need for awareness among clinicians of this disorder and the importance of examining for other associated clinical features to avoid missing such a vital diagnosis.
Case presentationA 69-year-old man of Chinese ethnicity presented to the Department of General Medicine with a 6-month history of left-sided neck swelling, associated with abdominal distension and progressive numbness and weakness in the hands and feet. He had weight loss and intermittent fever. He has a history of Type 2 diabetes mellitus which was diagnosed a year ago. He has no other significant medical or family history. He is married with three children. He is a non-smoker and a teetotaller. He works as a teacher.
On examination, he was alert but cachexic. He had palpable lymph nodes in his left cervical region, the largest measuring 4 × 4 cms. His abdomen was grossly distended with a positive fluid thrill.
He had undergone regular therapeutic abdominal paracentesis for the previous six months without the cause of the ascites being investigated appropriately. A recent left cervical lymph node biopsy (Figure 1A–E) was performed, which confirmed mixed-type Castleman Disease.
 Hematoxylin and Eosin × 40: The lymphoid follicles are increased in number. The mantle zones are expanded with lymphocytes giving rise to an onion skin-like aspect. Endothelial venule proliferation and hyalinization in the interfollicular zones are present. Sheets of plasma cells are seen in the interfollicular area. (B–E) Panel of immunohistochemical staining (×40 magnifications) shows positivity of CD 20, CD138, lambda light chain and kappa light chain restrictions. (F) Peritoneal fluid morphology shows a cluster of plasma cells with finely and coarsely vacuolated macrophages.")
(A) Hematoxylin and Eosin × 40: The lymphoid follicles are increased in number. The mantle zones are expanded with lymphocytes giving rise to an onion skin-like aspect. Endothelial venule proliferation and hyalinization in the interfollicular zones are present. Sheets of plasma cells are seen in the interfollicular area. (B–E) Panel of immunohistochemical staining (×40 magnifications) shows positivity of CD 20, CD138, lambda light chain and kappa light chain restrictions. (F) Peritoneal fluid morphology shows a cluster of plasma cells with finely and coarsely vacuolated macrophages.
He was referred to the Department of Hematology upon histological confirmation of Castleman disease. On further examination, he had flushing and generalized skin thickening with microstomia, which were consistent with sclerodermoid-like skin changes. He had a palpable liver and spleen. The neurological examination revealed a mixed sensorimotor peripheral neuropathy in both hands and feet in a glove and stocking distribution. The fundoscopy did not show any papilledema. He had bilateral pitting pedal edema extending to the mid shins bilaterally. Other systemic examinations were unremarkable.
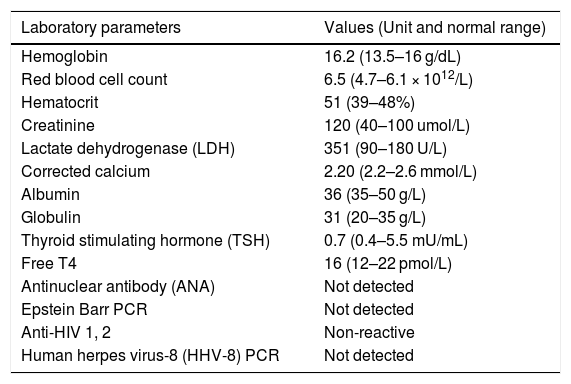
His complete blood count revealed erythrocytosis with normal white cell and platelet parameters. The other laboratory parameters are as tabulated in Table 1. Abdominal paracentesis revealed large-volume serous transudative ascites with the peritoneal fluid morphology (Figure 1F), showing a cluster of plasma cells with foamy macrophages. The chest radiograph portrayed bilateral pleural effusion. A Whole Body Computed Tomography (WBCT) imaging depicted multiple enlarged supra- and infra-diaphragmatic lymphadenopathies with the largest lymph node measuring 5 × 5 cms and multiple osteosclerotic bone lesions at T3, T9 and T12 vertebral bodies. A nerve conduction study confirmed symmetrical demyelinating polyneuropathy with diffuse axonal loss. Serum protein electrophoresis (SPEP) revealed a monoclonal M spike in the beta region. The serum M protein concentration was 9 g/L. The serum and urine immunofixation were positive for monoclonal gammopathy in the lambda region. Serum free light chain analysis (SFLC) demonstrated kappa light chain levels at 6.22 mg/L (normal range: 3.20–19.20 mg/L) and elevated lambda light chain levels at 2590 mg/L (normal range: 5.60–27.60 mg/L). The lambda/kappa ratio was 416. The bone marrow aspiration and trephine did not show any infiltration.
Tabulation of laboratory parameters.
| Laboratory parameters | Values (Unit and normal range) |
|---|---|
| Hemoglobin | 16.2 (13.5–16 g/dL) |
| Red blood cell count | 6.5 (4.7–6.1 × 1012/L) |
| Hematocrit | 51 (39–48%) |
| Creatinine | 120 (40–100 umol/L) |
| Lactate dehydrogenase (LDH) | 351 (90–180 U/L) |
| Corrected calcium | 2.20 (2.2–2.6 mmol/L) |
| Albumin | 36 (35–50 g/L) |
| Globulin | 31 (20–35 g/L) |
| Thyroid stimulating hormone (TSH) | 0.7 (0.4–5.5 mU/mL) |
| Free T4 | 16 (12–22 pmol/L) |
| Antinuclear antibody (ANA) | Not detected |
| Epstein Barr PCR | Not detected |
| Anti-HIV 1, 2 | Non-reactive |
| Human herpes virus-8 (HHV-8) PCR | Not detected |
In view of his presentation, he was diagnosed with the multicentric mixed-type Castleman disease variant of the POEMS syndrome. He was treated with six cycles of oral melphalan at 4 mg/m2 and dexamethasone at 20 mg per week. He was compliant to his treatment and did not develop any gross adverse effects from it. He was not suitable for autologous peripheral blood stem cell transplantation, as he was over the age of 65. His extravascular volume overload (pleural effusion and ascites) and neurological symptoms improved significantly. He did not require any further abdominal paracentesis. He is no longer cachexic and has no palpable lymph nodes. The repeated post-chemotherapy serum free light chain analysis showed no elevated light chains. He is on a regular three-monthly follow-up at the hematology clinic. He has been in complete remission for the past 12 months.
DiscussionWe report an interesting case of a Multicentric Mixed Pathology Castleman Disease variant of the POEMS syndrome in a patient who presented with persistent unexplained large-volume ascites. This diagnosis is often missed by many clinicians due to its rarity in clinical practice. The multicentric Castleman disease usually presents with constitutional symptoms and diffuse lymphadenopathy. It occurs in all age groups and is most frequently seen in the fourth and fifth decades of life, with a higher incidence in males.
The POEMS syndrome is characterized by polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy and skin manifestations. The diagnosis of POEMS syndrome includes two major mandatory criteria, one other major criteria and at least one minor criteria.4 The major mandatory criteria are demyelinating polyneuropathy and monoclonal plasma cell disorder, mainly of lambda predominance. The other major criteria include Castleman disease, osteosclerosis and elevated vascular endothelial growth factor (VEGF). Minor criteria include organomegaly, extravascular volume overload, endocrinopathy, skin changes, papilledema and thrombocytosis/polycythemia. Our patient fulfilled 3 major criteria and 5 minor criteria.
The pathophysiology of Castleman disease is associated with inflammatory hypercytokinemia, most notably elevated levels of interleukin-6 (IL-6) and the IL-6 receptor. The elevated IL-6 results in an overgrowth of B-lymphocytes and plasma cells which contribute to tumor proliferation.5 In addition, the extravascular volume overload, such as ascites, pleural effusion and peripheral edema manifested by our patient in this case, is most likely attributed to the elevated (VEGF), which contributes to tumor angiogenesis and enhanced vascular permeability. High IL-6 also augments the inflammatory reaction, producing a series of constitutional symptoms. No single gene has been identified, suggesting that the disease may be influenced by ethnicity and genetic factors, such as the polymorphism of the IL-6 receptor.6
In our case, we were unable to quantify the serum IL-6 and plasma VEGF levels, as these services were unavailable in the country.
Among the histological subtypes of multicentric Castleman disease, the plasma cell type predominates. However, other subtypes, such hyaline vascular and mixed pathology, are also seen. The hyaline vascular subtype is often described as having an onion skin appearance in which the follicles are surrounded by concentric layers of mantle cells and the presence of a radially penetrating sclerotic blood vessel (lollipop sign).7 The plasmacytic subtype usually shows an increased number of follicles with hyperplastic germinal centers and sheets of plasma cells. In our case, the cervical lymph node tissue demonstrated features of both hyaline vascular and plasma cell subtypes, thus resulting in a mixed pathology diagnosis.
Many regimens are used to treat the Castleman disease variant of the POEMS syndrome. Melphalan-dexamethasone has demonstrated an 81% haematologic response rate, with 100% showing some neurological improvement.8 Another treatment regimen used with a good response is lenalidomide/dexamethasone. Cyclophosphamide/dexamethasone resulted in at least 50% of patients having clinical improvement.8 High-dose therapy using melphalan, ranging from 140–200 mg/m2, followed by autologous peripheral blood stem cell transplantation, remains the first-line treatment for patients younger than 65 years of age with normal organ function.9 It is effective, with 100% of patients showing some neurological improvement.9
ConclusionThis case makes an interesting observation of the Castleman disease variant of the POEMS syndrome in a patient who presented repeatedly with worsening ascites. Hence, it is always imperative to examine for other associated clinical features to avoid missing such a vital diagnosis. The POEMS syndrome progresses rapidly without therapy and may become life-threatening if the diagnosis is delayed.
ConsentWritten informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Author’s contributionBoth authors contributed equally to the production of this manuscript.
Conflicts of interestThe authors declare no conflicts of interest.


