
Older patients with acute myeloid leukemia are particularly difficult to treat, as they have a high risk of comorbidities, poor performance status and less tolerability to chemotherapy, as well as a more aggressive disease biology, responsible for the resistance to treatment. There is a need to explore novel therapeutic agents that are more effective and tolerable. Venetoclax, a BCL-2 inhibitor is a promising agent, as BCL-2 overexpression is present in 84% of acute myeloid leukemia patients at diagnosis and 95% of patients at relapse and has been associated with leukemia cell survival, chemotherapy resistance and poor prognosis.
ObjectiveTo review the available data about venetoclax in acute myeloid leukemia and how it can influence the treatment in older patients.
MethodsUsing the Pubmed database, we selected 29 articles published within the last 15 years, considering preclinical and clinical trials and review studies that combined venetoclax with acute myeloid leukemia.
ResultsVenetoclax has demonstrated promising results in preclinical and clinical trials, especially in patients with poor prognosis and the IDH mutation, with an excellent side-effect profile. However, resistance seems to develop rapidly with venetoclax monotherapy, because of antiapoptotic escape mechanisms.
ConclusionsWhile the results with the use of venetoclax seem encouraging, it is not likely that targeting a single pathway will result in long-term disease control. The solution includes the use of combined therapy to block resistance mechanisms and enhance apoptosis, by reducing MCL-1, increasing BIM or inhibiting the complex IV in the mitochondria.
Acute myeloid leukemia (AML) is a hematopoietic neoplasm involving precursor cells committed to the myeloid lineage of cellular development that faces a series of genetic changes, which results in clonal proliferation of myeloid precursors with impaired capacity to differentiate into normal mature cells.1,2 This results in an accumulation of leukemic blasts or immature forms in the bone marrow, peripheral blood and other tissues and reduction of normal cells, expressed as systemic signs, such anemia, bleeding and increased risk of infection.1
AML represents 80 percent of acute leukemia in adults .1 and annually, more than one quarter of a million adults throughout the world are diagnosed with AML,3 with 60% being over 60 years of age.4 . Although it represents the most common type of leukemia in adults, it continues to have the lowest survival rate of all.5 In fact, two-thirds of young adults and 90% of older adults still die of this disease,3 with only 20% of older patients surviving 1 year.6
That is why AML remains a challenge to both patients and clinicians. In fact, despite advances in our understanding of the AML pathogenesis, classification, genomic, and prognostic factors, treatment minimally changed in the last 40 years and outcomes remain poor for the majority of older patients.7
The disappointing outcomes in older patients are explained not only by a poor performance status and a higher prevalence of comorbidities, which make intensive chemotherapy more toxic and less tolerated, but also by adverse disease characteristics, such as unfavorable cytogenetics (−7/del(7), −5/del(5q) or 5p, inv(3)/t(3;3), t(6;9), complex karyotype, monosomal karyotype, 12p, 17p, and 11q23/mixed-lineage leukemia aberrations, excluding t(9;11)) and unfavorable gene mutations (DNMT3A, ASXL1, RUNX1 and TP53 mutations), higher incidence of secondary AML, including AML with prior myelodysplasia and therapy-related AML, and multidrug resistance (MDR) phenotypes, determined by the expression of MDR1 (or p-glycoprotein) in 71% of older AML patients, higher prevalence of the CD34+ phenotype (65%), and higher rates of drug efflux (58%), which make the leukemic cells more resistant to chemotherapy.4,6,8–14
Moreover, cells from older patients with AML have lower apoptotic rates in culture than those of younger patients with AML, and lower rates of apoptosis after exposure to chemotherapy.6 In fact, overexpression of the anti-apoptotic B cell leukemia/lymphoma-2 (BCL-2) gene is upregulated in 84% of AML patients at diagnosis and 95% at relapse,15 and is associated with poor prognosis, lower complete remission (CR) rate, significantly shorter survival,2,16 AML cell survival and chemotherapy resistance.17
Therefore, older adults have lower rates of CR and only 38–62% of patients over 60 years of age with de novo AML achieve CR, compared to 65–73% of total patients, with standard induction therapy.16
There is a need for newer therapies and a more individualized approach for the treatment of AML.18 Recently, thanks to a better knowledge of the molecular pathogenesis of AML, there have been an increasing number of potential targets and pathways that can be used for specific targeted therapy in AML.7,19
The BCL-2 family regulates the mitochondrial pathway of apoptosis.20,21 The balance between pro-apoptotic BH3-only proteins and anti-apoptotic BCL-2 proteins determines the life or death of a cell.21 Agents that inhibit the anti-apoptotic BCL-2 family proteins bind to the BH3 binding groove, mimicking the BH3 domain of BH3-only proteins, thereby liberating the proapoptotic proteins BCL-2 antagonist/killer 1 (BAK) and BCL-2-associated X protein (BAX) to trigger apoptosis, and are designated as BH3 mimetics.18,22,23 Earlier investigational BH3 mimetics were found to bind efficiently to several antiapoptotic proteins, such as the BCL-2, B-cell lymphoma-extra large (BCL-XL) and the myeloid cell leukemia sequence 1 (MCL-1), but were associated with on-target toxicity and thrombocytopenia because platelets depend on the BCL-XL for their survival.18 That is the case of navitoclax (ABT-263), a BCL-2 and BCL-XL inhibitor. However, the clarification of the mechanism by which navitoclax causes thrombocytopenia suggested that a more selective BCL-2 inhibitor could prevent this toxicity and enable a higher dosing to increase clinical efficacy. This led to the rational reverse engineering of navitoclax to create venetoclax (ABT-199).24,25
Treatment with venetoclax seems suitable in cancers with the marked overexpression of BCL-2,18,21–23 such as AML, and may be useful in increasing the apoptotic response and improve clinical outcomes.
In this study, we will review the available data on venetoclax in AML and how it can influence the treatment of AML in older patients.
MethodsIn this review we used the database and searched for the Medical Subject Heading (MeSH) terms (“BCL-2 inhibitor” OR “Venetoclax” OR “ABT-199”) AND (“acute myeloid leukemia” OR “AML”). The limits considered were the English language and articles published within the last 15 years. The final update of the search was conducted in February 2018.
In the selection of the articles, we analyzed the title and the abstract, followed by the full text of the previously selected studies, and excluded those which were not related to the theme, considering preclinical and clinical trials and review studies that combined acute myeloid leukemia with venetoclax or ABT-199.
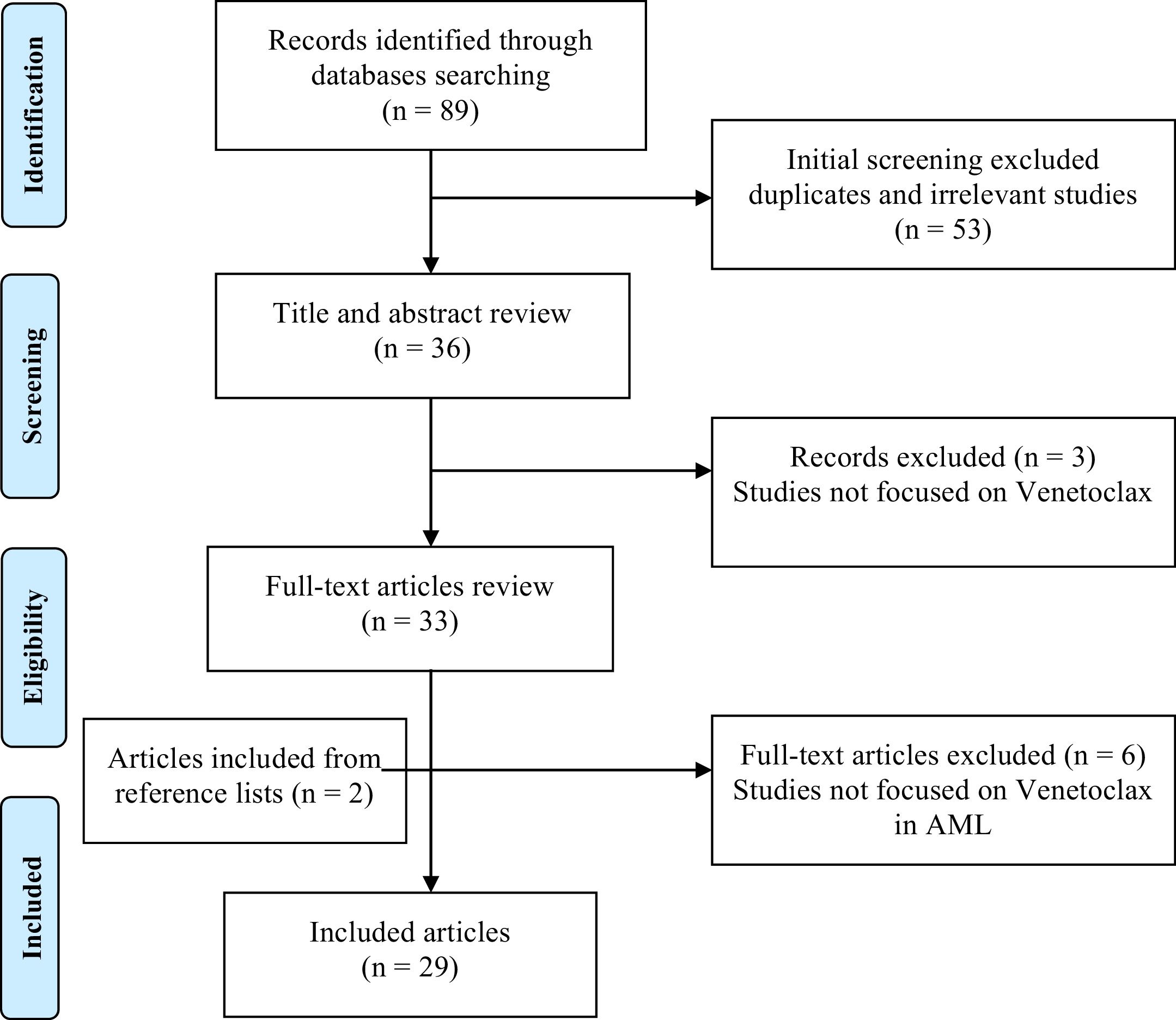
Additional articles were further investigated through a manual search in relevant reference lists. The detailed search strategy is presented in Figure 1.

Overall, this research resulted in a total of 29 articles, which comprehends the literary basis of this review.
ResultsVenetoclax (ABT-199)Mechanism of actionVenetoclax (ABT-199), a more advanced BH3 mimetic, is an orally bioavailable BCL-2-specific inhibitor that selectively inhibits BCL-2, but not BCL-XL, and thus does not cause thrombocytopenia, and may be useful in malignancies in which BCL-2 plays a more central role than BCL-XL, such as AML. It has an excellent side-effect profile and, compared to navitoclax, has a slightly higher avidity for the BCL-2 and three orders of magnitude less avidity for the BCL-XL.18,22–25
The AML blasts and AML stem cells are dependent on the BCL-2 for survival, while normal hematopoietic cells are dependent on the MCL-1.18 Hence, importantly, the BCL-2 inhibition relatively spares normal hematopoietic stem cells, which are more dependent on the MCL-1 for their survival.26
Venetoclax-based therapy could provide significant survival prolongation for older patients with AML who are ineligible for more aggressive therapies and, therefore, has the potential to revolutionize the AML therapy.25
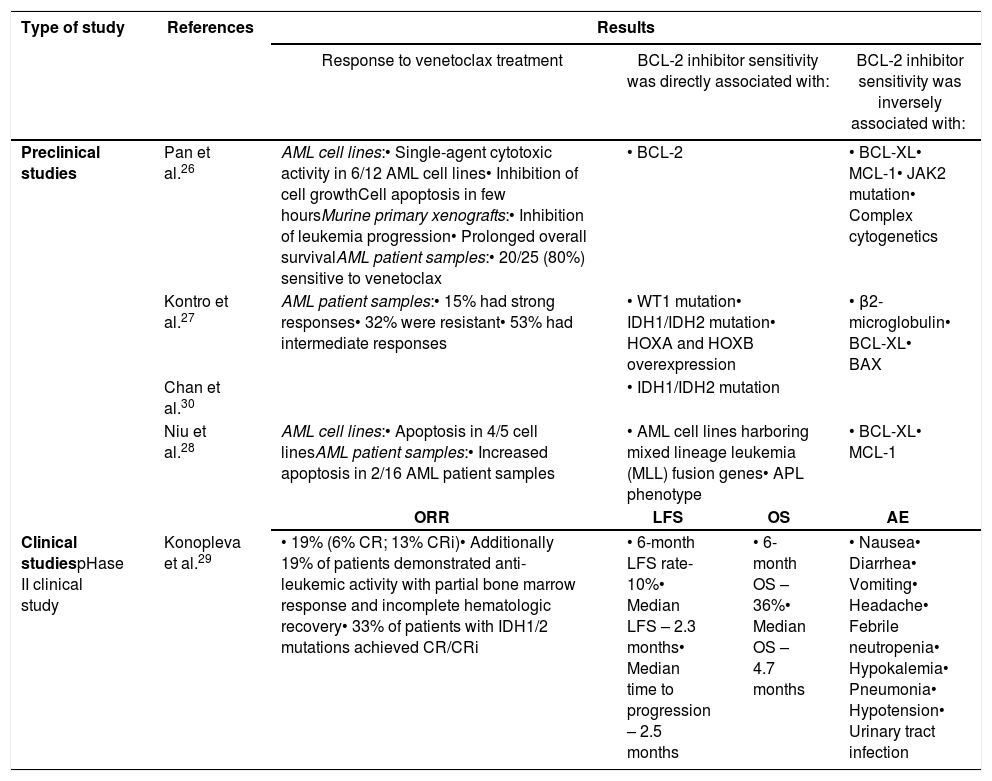
Preclinical studiesIn a preclinical study, Pan et al.26 tested the activity of venetoclax in leukemia cell lines, murine primary xenografts and primary samples of patients with AML. This study demonstrated significant activity of venetoclax, as a single agent, in all three AML model systems (Table 1).18,26 In a sensitive AML cell line, cell growth was inhibited and cell apoptosis was observed within just 2h of exposure to venetoclax. In vivo exposure to venetoclax, on murine primary xenografts, showed inhibition of leukemia progression and prolonged overall survival (OS). Furthermore, in primary patient AML cells, including AML cells with diploid cytogenetics and mutations in FMS-like tyrosine kinase-3 (FLT3), NRAS, and nucleophosmin (NPM1) genes, 20 out of 25 (80%) were sensitive to venetoclax, while 5 samples were resistant.
Venetoclax in monotherapy.
| Type of study | References | Results | |||
|---|---|---|---|---|---|
| Response to venetoclax treatment | BCL-2 inhibitor sensitivity was directly associated with: | BCL-2 inhibitor sensitivity was inversely associated with: | |||
| Preclinical studies | Pan et al.26 | AML cell lines:• Single-agent cytotoxic activity in 6/12 AML cell lines• Inhibition of cell growthCell apoptosis in few hoursMurine primary xenografts:• Inhibition of leukemia progression• Prolonged overall survivalAML patient samples:• 20/25 (80%) sensitive to venetoclax | • BCL-2 | • BCL-XL• MCL-1• JAK2 mutation• Complex cytogenetics | |
| Kontro et al.27 | AML patient samples:• 15% had strong responses• 32% were resistant• 53% had intermediate responses | • WT1 mutation• IDH1/IDH2 mutation• HOXA and HOXB overexpression | • β2-microglobulin• BCL-XL• BAX | ||
| Chan et al.30 | • IDH1/IDH2 mutation | ||||
| Niu et al.28 | AML cell lines:• Apoptosis in 4/5 cell linesAML patient samples:• Increased apoptosis in 2/16 AML patient samples | • AML cell lines harboring mixed lineage leukemia (MLL) fusion genes• APL phenotype | • BCL-XL• MCL-1 | ||
| ORR | LFS | OS | AE | ||
| Clinical studiespHase II clinical study | Konopleva et al.29 | • 19% (6% CR; 13% CRi)• Additionally 19% of patients demonstrated anti-leukemic activity with partial bone marrow response and incomplete hematologic recovery• 33% of patients with IDH1/2 mutations achieved CR/CRi | • 6-month LFS rate-10%• Median LFS – 2.3 months• Median time to progression – 2.5 months | • 6-month OS – 36%• Median OS – 4.7 months | • Nausea• Diarrhea• Vomiting• Headache• Febrile neutropenia• Hypokalemia• Pneumonia• Hypotension• Urinary tract infection |
AML: acute myeloid leukemia; ORR: overall response rate; LFS: leukemia free survival; OS: overall survival; AE: adverse events; CR: complete remission; CRi: complete remission with incomplete blood count recovery.
Kontro et al.27 evaluated the ex vivo sensitivity of fresh leukemic cells from 73 diagnosed and relapsed/refractory AML patients, subsequently analyzing if the responses correlated to specific mutations or gene expression. The strongest responses were observed in 15% of the AML patient samples, 32% were resistant, and the remaining presented intermediate responses to venetoclax (Table 1).
Another study, developed by Niu et al.,28 concluded that venetoclax was able to induce apoptosis in a dose-dependent manner in four of the five cell lines tested. Similar to the cell line results, venetoclax was able to induce a dose-dependent increase in apoptosis in two AML patient samples (Table 1).28
Overall, in preclinical studies, venetoclax has shown potent antileukemic activity in AML cell lines and primary patient samples in vitro and in murine xenograft models.25,29 These data provide rationale for targeted BCL-2 inhibition with venetoclax in AML in clinical trials.25,29
Clinical studiesIn a phase II clinical study,29 to evaluate the efficacy of single-agent venetoclax in patients with high-risk relapsed/refractory AML or in those unfit for intensive chemotherapy, 32 patients were evaluated and their overall response rate (ORR) by the International Working Group (IWG) was 19%, with 6% achieving a CR and 13% achieving a CR with incomplete blood count recovery (CRi). Except for 1 CRi, all objective responses were achieved by the week 4 assessment and all IWG-defined responses were observed in patients who had been previously treated for AML (Table 1).29
Additionally 19% of patients demonstrated anti-leukemic activity not meeting IWG criteria, with partial bone marrow response and incomplete hematologic recovery.29
Also, venetoclax appeared to be safe and well-tolerated, with a similar range of adverse events (AEs) seen in other studies, with 84% presenting severe AEs, being febrile neutropenia the most common (31%). These AEs were consistent with expectations in this population. There were no events of tumor lysis syndrome (TLS) and no AEs leading to death.29
Such results, although seemingly limited, have not been observed with any other oral monotherapy for AML and are particularly impressive considering that their cohort consisted of older patients and included many patients with high-risk features.25 In fact, most patients had pre-existing hematologic disorders or myeloproliferative neoplasms (41%), therapy-related AML with complex cytogenetics (13%) and older age (median 71 years). Also, these patients were heavily pre-treated, 41% having progressed through at least three prior treatments, 53% having received standard “7+3″ induction therapy (cytarabine+anthracycline) and 75% having failed hypomethylating agents.29
Moreover, mutations in isocitrate dehydrogenase (IDH) 1 or 2 were identified in 38% of patients. Among the patients with IDH1/2 mutations, 33% achieved CR/CRi and several other patients with IDH mutations had significant blast count reduction that did not meet IWG response criteria.29 These clinical observations confirm in vitro observations that suggest an increased sensitivity of IDH-mutated AML to BCL-2 inhibition.18,30
None of the patients with therapy-related AML experienced anti-leukemic activity on venetoclax.29
The 6-month leukemia-free survival (LFS) rate was 10% and the median LFS was 2.3 months. The 6-month OS estimate was 36% and median OS was 4.7 months.29
In summary, relapsed/refractory AML in older patients has an extremely poor prognosis, with OS less than 6 months. So, a response rate of 19% plus additional cases of myeloblast reduction, with a relatively well tolerated oral single agent, is a promising clinical accomplishment.29 However, all patients treated with venetoclax relapsed, despite the potential initial response, with a median time to progression of 2.5 months. Therefore, resistance appears to develop rapidly with venetoclax monotherapy.25
Predictors of responsePredictive biomarkers may be used to identify and select patients more responsive to venetoclax therapy, therefore potentially improving the response rate.29
In the preclinical study of Pan et al.,26 the cell lines more sensitive to venetoclax were those with high levels of BCL-2 protein and relatively low BCL-XL and MCL-1 expression. Similar results were found in primary AML myeloblasts. Moreover, AML myeloblasts demonstrated higher BCL-2 mRNA expression than normal bone marrow. Samples from patients with complex cytogenetics and janus kinase 2 (JAK2) mutation were largely insensitive to venetoclax and no correlation was found between venetoclax sensitivity and French-American-British (FAB) classification (Table 1).26
The in vitro results suggested that there will be heterogeneity in clinical response, so a predictive biomarker would be of great utility. In this context, they presented four methods that may be predictive of clinical response to venetoclax. The first method is cytogenetics, cellular death response to venetoclax appears to be largely independent of cytogenetic and genetic mutation status, except for complex karyotype and JAK2 mutation that are resistant, suggesting that treatment with venetoclax could be useful for a large proportion of patients with poor prognostic factors. A second method is an ex vivo short-term culture of the primary patient samples with venetoclax. The third predictive biomarker method is to measure BCL-2 levels by Western blotting, as they showed that increased expression of BCL-2 is associated with increased sensitivity to venetoclax. The fourth method, BH3 profiling, that is a functional assay which accounts for the relative amounts and interactions of all of the BCL-2 family members, can function as a predictive biomarker since it could discriminate in vivo sensitivity of human AML cells to venetoclax.26 Therefore, BH3 profiling of pre-treatment AML samples can be used to identify and select AML cases that are mostly BCL-2 dependent and so more responsive to venetoclax treatment, potentially improving the response rate.26,29
To also identify robust biomarkers for BCL-2 inhibitor sensitivity, Kontro et al.27 developed a preclinical study concluding that BCL-2 inhibitor sensitivity was associated with genetic aberrations in chromatin modifiers, wilms tumor 1 (WT1) and IDH, homeobox (HOX) A and B gene transcripts overexpression and BAX expression. Significant inverse correlation responses to venetoclax were related to β2-microglobulin and in a lesser degree to BCL-XL expression (Table 1).27
A study of Chan et al.30 also showed that IDH1- and IDH2-mutant primary human AML cells were more sensitive than IDH1/2 wild-type cells to venetoclax both ex vivo and in xenotransplant models (Table 1). This has been attributed to suppression of complex IV, also referred to as cytochrome c oxidase, in the mitochondrial electron transport chain (ECT) by the oncometabolite 2-hydroxyglutarate (2-HG), generated by IDH mutations, that lowers the mitochondrial threshold to trigger apoptosis upon BCL-2 inhibition. The 2-HG accumulation in AML cells with the IDH1/2 mutation directly inhibits the complex IV and thus, increases the dependency on BCL-2 to prevent apoptosis. Hence, the IDH1/2 mutation status may identify patients that are likely to respond to pharmacologic BCL-2 inhibition, considering that approximately 15% of AML patients have mutations in either IDH1 or IDH2.30 However, the study of Chan et al.30 showed that complex IV inhibition is sufficient to sensitize AML cells to apoptosis, upon venetoclax treatment, and offers a novel strategy to enhance clinical efficacy and overcome potential resistance in patients with unmutated IDH.30,31
Another study, developed by Niu et al.,28 whose objective was to identify genetic subgroups which may better respond to venetoclax and biomarkers predicting venetoclax sensitivity in AML, concluded that AML cell lines harboring mixed lineage leukemia (MLL) fusion genes were especially sensitive to venetoclax (Table 1). Rearrangements in the MLL gene on chromosome 11q23 occur in up to 10% of AML and are generally associated with a relatively unfavorable prognosis. Resistance to therapy for AML in general, and MLL leukemias in particular, may also stem from the aberrant expression of BCL-2 family proteins.28
Patient samples with the acute promyelocytic leukemia (APL) phenotype were significantly more sensitive to single-agent venetoclax. Moreover, in the combined cohort of AML cell lines and patient samples were the overexpression of BCL-XL and MCL-1 attenuated venetoclax-induced apoptosis. These suggest that the ratios of BCL-2/MCL-1 transcripts may represent a good biomarker predicting venetoclax sensitivity in AML.28
Mechanisms of resistance to venetoclaxAlthough venetoclax is the first BCL-2-selective inhibitor that has demonstrated promising results in multiple cancers, including AML, intrinsic drug resistance remains a concern.32,33 In fact, venetoclax has demonstrated limited efficacy in BCL-XL and MCL-1-dependent malignancies. Niu et al.,34 Choudhary et al. and Lin et al.,35 demonstrated that intrinsically resistant AML cells have increased levels of MCL-1 and BCL-XL following venetoclax treatment, resulting in increased sequestration of BCL-2-like 11 (BIM) by MCL-1.34,35
Venetoclax treatment was found to result in dissociation of pro-apoptotic BIM from BCL-2, demonstrating its role in inducing apoptosis in AML cells, but also resulted in increased sequestration of BIM by MCL-1, preventing BIM from inducing apoptosis, while also resulting in increased MCL-1 protein levels in venetoclax-resistant AML cells. These results suggest that MCL-1 is a key player in the intrinsic resistance to venetoclax in AML cells. Since venetoclax does not inhibit MCL-1, increased expression of MCL-1 could be a potential source of upfront resistance to BCL-2 inhibition by venetoclax. Confirming this, the CRISPR knockdown of MCL-1 significantly enhanced venetoclax-induced apoptosis in AML cells.26,32,33
Therefore Lin et al.,35 demonstrated that by targeting MCL-1 and BCL-XL, resistant AML cell lines could be resensitized to venetoclax. Furthermore, targeting MCL-1 and/or BCL-XL along with administration of venetoclax was capable of delaying or preventing the acquisition of drug resistance.35
Niu et al.34 proved also that this mechanism of resistance can be overcome by combining venetoclax with DNA damaging agents daunorubicin or cytarabine, which reduce MCL-1 levels and enhance venetoclax activity in venetoclax-resistant AML cells. It is plausible that in the venetoclax-sensitive cells more BIM is released from BCL-2 than can be sequestered by MCL-1 (high BCL-2/MCL-1 transcript ratio), resulting in free BIM, which would then activate the apoptosis pathway, while in venetoclax-resistant cells (low BCL-2/MCL-1 transcript ratios) it is likely that there is enough MCL-1 to sequester BIM and inhibit apoptosis. For the combined drug treatments in resistant cells, MCL-1 downregulation likely results in free BIM, allowing for activation of BAK/BAX and triggering of apoptosis.34 Additionally, they demonstrated that these combinations are synergistic in cell lines and primary patient samples independent of their sensitivities to venetoclax, thus providing evidence that screening for venetoclax resistance is not necessary, and therefore supporting clinical testing regardless of venetoclax sensitivity.34
Overall, it is unlikely that any single approach, including the most targeted, will be successful in eradicating AML cells, due to the related problems of intrinsic or acquired forms of resistance. One approach to circumventing this problem involves combining agents that act in complementary ways to promote cell death or to suppress cellular escape mechanisms that facilitate the acquisition of drug resistance.7,35
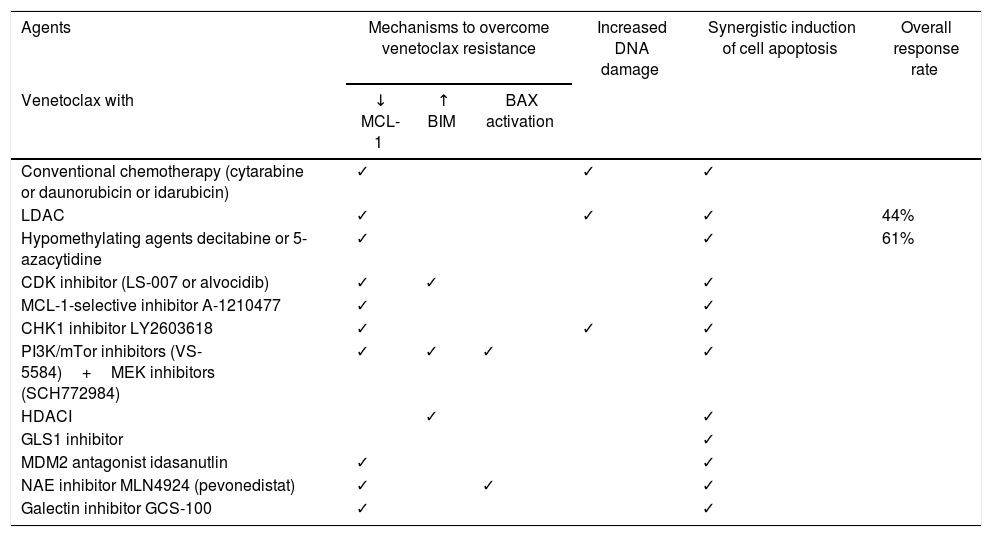
Combined therapyOnce MCL-1 has been related to intrinsic resistance to venetoclax in AML cells, drugs that act via MCL-1 inhibition are being explored, as a manner in which to overcome this resistance mechanism. Decreased MCL-1 protein levels were demonstrated in preclinical studies in the combination of venetoclax with chemotherapeutic agents cytarabine and/or daunorubicin or idarubicin, hypomethylating agents decitabine or 5-azacytidine, CDK9 inhibitors LS-007 and alvocidib, MCL-1 selective inhibitor A-1210477, Checkpoint kinase 1 (CHK1) inhibitor LY2603618, PI3K/mTOR inhibitors VS-5584 plus MEK inhibitors SCH772984, Mouse double minute 2 homolog (MDM2) antagonist idasanutlin, Nedd8 activating enzyme (NAE) inhibitor MLN4924 (pevonedistat) and Galectin inhibitor GCS-100 (Table 2).24,32,36–42
Combined therapy.
| Agents | Mechanisms to overcome venetoclax resistance | Increased DNA damage | Synergistic induction of cell apoptosis | Overall response rate | ||
|---|---|---|---|---|---|---|
| Venetoclax with | ↓ MCL-1 | ↑ BIM | BAX activation | |||
| Conventional chemotherapy (cytarabine or daunorubicin or idarubicin) | ✓ | ✓ | ✓ | |||
| LDAC | ✓ | ✓ | ✓ | 44% | ||
| Hypomethylating agents decitabine or 5-azacytidine | ✓ | ✓ | 61% | |||
| CDK inhibitor (LS-007 or alvocidib) | ✓ | ✓ | ✓ | |||
| MCL-1-selective inhibitor A-1210477 | ✓ | ✓ | ||||
| CHK1 inhibitor LY2603618 | ✓ | ✓ | ✓ | |||
| PI3K/mTor inhibitors (VS-5584)+MEK inhibitors (SCH772984) | ✓ | ✓ | ✓ | ✓ | ||
| HDACI | ✓ | ✓ | ||||
| GLS1 inhibitor | ✓ | |||||
| MDM2 antagonist idasanutlin | ✓ | ✓ | ||||
| NAE inhibitor MLN4924 (pevonedistat) | ✓ | ✓ | ✓ | |||
| Galectin inhibitor GCS-100 | ✓ | ✓ | ||||
Another approach would be to upregulate BIM levels, leading to an increased BIM/MCL-1 ratio favoring apoptosis. Increased BIM levels were demonstrated in preclinical studies in the combination of venetoclax with CDK9 inhibitor alvocidib, PI3K/mTOR inhibitors VS-5584 plus MEK inhibitors SCH772984 and Histone deacetylase inhibitors (HDACI) (panobinostat) (Table 2).33,37,39
All the combinations described above, plus the combination of venetoclax with GLS1 inhibitor, resulted in synergistic induction of apoptosis in preclinical studies (Table 2),16,32–34,36–45 suggesting potential novel therapeutic strategies for AML. However, the biggest challenge is choosing the most promising combinations and bringing them forward into clinical trials, which are costly and time-consuming.20
Until now, clinical studies combining venetoclax were conducted with low-dose cytarabine (LDAC) and hypomethylating agents. In a phase 1b/2 study,44 with 18 older patients with AML, not eligible for intensive chemotherapy, venetoclax combined with LDAC achieved an ORR of 44% (CR of 22% and CRi of 22%). Tolerability was acceptable, with febrile neutropenia being the most common severe AE (33.3%). No significant TLS was observed. Two deaths occurred.17,25,44
In a phase Ib clinical trial,46 with a cohort of 57 older patients with AML ineligible for standard chemotherapy, venetoclax combined with hypomethylating agents decitabine or 5-azacytidine achieved an ORR of 61% (CR of 24% and CRi of 37%), without differences in response between patients who received decitabine or 5-azacitidine. Poor-risk cytogenetics was presented in 38% of patients, with an ORR of 52%. The IDH1/2 mutations were reported in 31% of patients, with an ORR of 59%. These drug combinations were well tolerated, with febrile neutropenia being the most frequent severe AE (31%). No events of TLS were reported. Twenty-six deaths occurred, 10 from adverse events. Median duration of response was 8.4 months with a median OS of 12.3 months.46
Discussion and conclusionVenetoclax has demonstrated promising results in preclinical and clinical trials, especially in patients with poor prognosis, and with an excellent side-effect profile. However, the response to venetoclax is heterogeneous, with sensitivity to the BCL-2 inhibitor being directly associated with BCL-2 and HOX A/B overexpression, BAX expression and WT1 and IDH1/2 mutation, and inversely associated with expression of MCL-1, BCL-XL, JAK2 mutation, β2-microglobulin and complex cytogenetics.
Indeed, cells with intrinsic or acquired resistance to venetoclax exhibited increased MCL-1 and BCL-XL levels, leading to increased sequestration of BIM. Unfortunately, resistance mechanisms seem to develop rapidly with venetoclax monotherapy, so, targeting MCL-1 and/or BCL-XL, along with the administration of venetoclax, has the capacity to delay or prevent the acquisition of drug resistance. However, BCL-XL inhibition is not a good strategy because it was associated with on-target toxicity and thrombocytopenia. On the other hand, MCL-1 is not inhibited by venetoclax, and increased MCL-1 is an important antiapoptotic escape mechanism. Therefore, MCL-1 inhibition seems to be an important approach to overcome venetoclax resistance, as well as to promote the upregulation of BIM.
Concerning the IDH mutation, its prevalence in the analyzed studies (38% in the study of Konopleva et al.29 and 31% in the study of DiNardo et al.46) raised the possibility that this mutation was more prevalent in older patients with AML, taking into account that Chan et al.,30 Stock et al.2 and Schiffer et al.1 refer a regular prevalence of 15%. However, these studies correspond to a small number of patients, and Tsai et al.,14 analyzing gene mutations in a larger cohort of AML patients (462 patients), reported a prevalence of the IDH1 mutation in 6.8% and IDH2 mutation in 14.7% of older patients. Moreover, Saygin et al.17 reported a prevalence range of 5–10% in the IDH1 mutation and 10–15% in IDH2 mutation and therefore, in the study of Tsai et al.,14 the percentages of the IDH 1/2 mutation in older patients were within the normal range. Hence, a higher prevalence of the IDH mutation in older patients with AML was not proved. However, it is clear that patients with the IDH 1/2 mutation demonstrated superior responses with the venetoclax treatment, firmly indicating its use in patients with the IDH mutation.
Taking into account that in these cases the use of IDH inhibitors may also be indicated, we raised the possibility of a combined therapy with venetoclax and IDH inhibitors, to enhance clinical responses. However, in theory, that may result in increased resistance, since the production of the oncometabolite 2-HG, proportionated by the mutation in the IDH, suppresses the complex IV, lowering the mitochondrial threshold and triggering apoptosis upon the BCL-2 inhibition, and the IDH inhibitors diminish the oncometabolite 2-HG, so it will also reduce complex IV inhibition and therefore the sensitivity to venetoclax. Therefore, in theory, combined therapy with venetoclax and IDH inhibitors may be not indicated, unless there is a third component that inhibits complex IV. Nevertheless, venetoclax could be used in a complementary way, in patients who demonstrated prior resistance to treatment with the IDH inhibitors.
Given this perspective, in patients with unmutated IDH, Chan et al.30 defend that complex IV inhibition is sufficient to sensitize AML cells to apoptosis upon venetoclax treatment, so the combined use of complex IV inhibitors could be a possibility to enhance the response to venetoclax. Complex IV inhibition mimics a state of oxygen deprivation, which can activate BAX/BAK to trigger apoptosis, if not antagonized by BCL-2. Treatment with venetoclax disrupts the complex formation between BCL-2 and BAX/BAK and, consequently, promotes the activation of proapoptotic proteins, leading to apoptosis.30 In this context, tigecycline, an anti-microbial agent of the glycylcycline class, currently used as a broad-spectrum antibiotic, is a promising agent in the treatment of AML, functioning as an inhibitor of subunits 1 and 2 of complex IV.47
In a preclinical trial, tigecycline selectively killed AML cells at all stages of development, as well as showing anti-leukemic activity in mouse models of human leukemia, while sparing normal hematopoietic cells. Leukemic cells owe their heightened sensitivity to tigecycline due to an increased dependence on mitochondrial function with enhanced basal oxygen consumption, compared to normal hematopoietic cells, demonstrating that the mechanism of tigecycline-mediated lethality is mitochondrial translation inhibition.47
In a phase 1 study, with 27 patients (median age 70 years) with relapsed and refractory AML, tigecycline administration showed a favorable safety profile, but none of the patients had evidence of disease response.48 Although, the authors try to justify these results as a lack of effective steady-state levels of tigecycline, it could also be due to an increased dependency on BCL-2, preventing apoptosis and thus, combining tigecycline with venetoclax could enable a superior clinical efficacy.
Therefore, the use of venetoclax has the potential to improve currently available therapies, but, while the results with the use of venetoclax seem encouraging, it is not likely that targeting a single pathway will result in long-term disease control. The solution includes the use of combined therapy to block resistance mechanisms and enhance apoptosis, by reducing MCL-1, increasing BIM or inhibiting complex IV in the mitochondria.
In this context, larger and longer clinical trials, combining venetoclax with LDAC and hypomethylating agents, as well as with tigecycline, could supply the results needed to integrate the use of combined therapy with venetoclax as a treatment option in older patients with AML, ineligible for conventional chemotherapy.
Conflicts of interestThe authors declare no conflicts of interest.


