
Dermatologic problems represent about 15–20% of visits to acute care settings.1 Skin manifestations are common in haematology patients and can result from infection, malignancy, or chemotherapy. Lymphoma is the seventh most common type of malignancy in both sexes. It is a neoplastic proliferation of lymphoid cells at various stages of differentiation and affects lymph nodes with infiltration into the bone marrow, spleen and thymus. However, extra nodal involvement is frequently seen in many cases.
The skin is one of the most frequent sites of extranodal involvement and vary in different series published. Data from large series reported in the literature have shown gastrointestinal (GI) tract, skin, bone, and brain to be the most common sites of extranodal lymphoma.2,3 There is substantial geographical variation as well.4 Also, it is important to distinguish primary cutaneous lymphoma from skin involvement in systemic lymphomas, as primary cutaneous B-cell lymphomas are a heterogeneous group of mature B-cells neoplasms with tropism for the skin, whose biology and clinical course differ significantly from the equivalent nodal lymphomas. Cutaneous T-cell lymphomas have an indolent clinical course; prompt and accurate diagnosis should be the rule. Up to 50% of patients have cutaneous involvement, which arises before the diagnosis in 79% of cases, concurrently in 8% and after the diagnosis in 13%.5
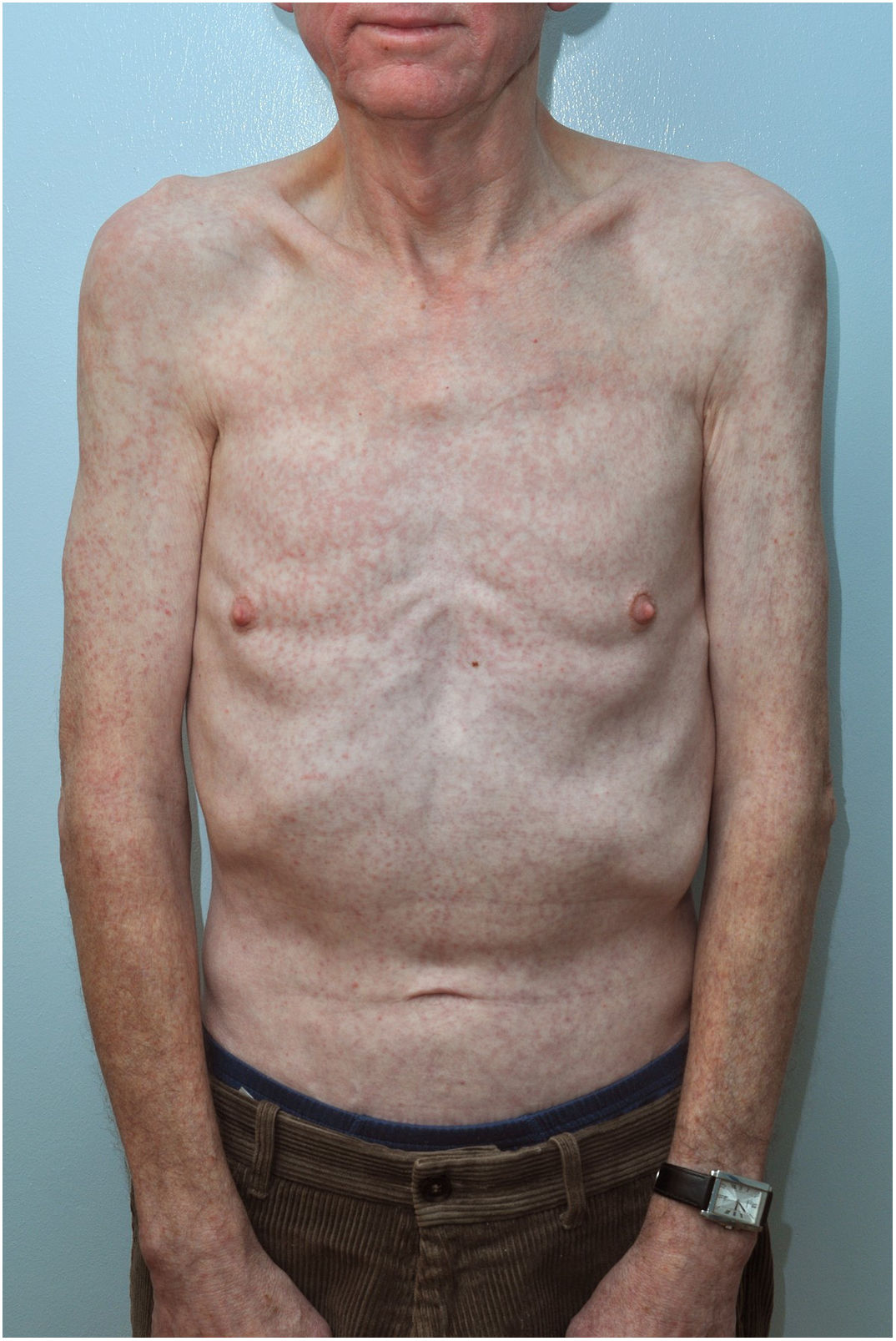
Case reportWe are presenting a case of a 64-year-old gentleman, with no significant past medical history until September 2017 when he initially presented to his GP with a 2 weeks history of generalized fatigue, body aches, arthritis and fever. Fever was high grade with rigours and chills, relieved by acetaminophen only to recur after 4h associated with dry cough and night sweats. He also complained of decreased appetite and some unintentional weight loss. There was complaint of generalized joint pains with mild swelling. His GP found few palpable cervical nodes and gave him a week course of Clarithromycin to help with his symptoms. He developed a generalized rash so stopped the medication and was admitted to our acute medical unit. He had mild cervical lymphadenopathy (improving) and non-tender hepatomegaly. His joints were slightly swollen with tenderness. There was a generalized maculopapular rash mainly on the trunk (Figure 1). His initial blood tests were unremarkable except for CRP 18 and ALT 58. CMV IgG was positive. EBV IgM and IgG were positive with EBV DNA 12570copies/mL. A viral serology for HBsAg, anti-HCV and anti-HIV were negative. Blood cultures were sterile. An echocardiography was normal

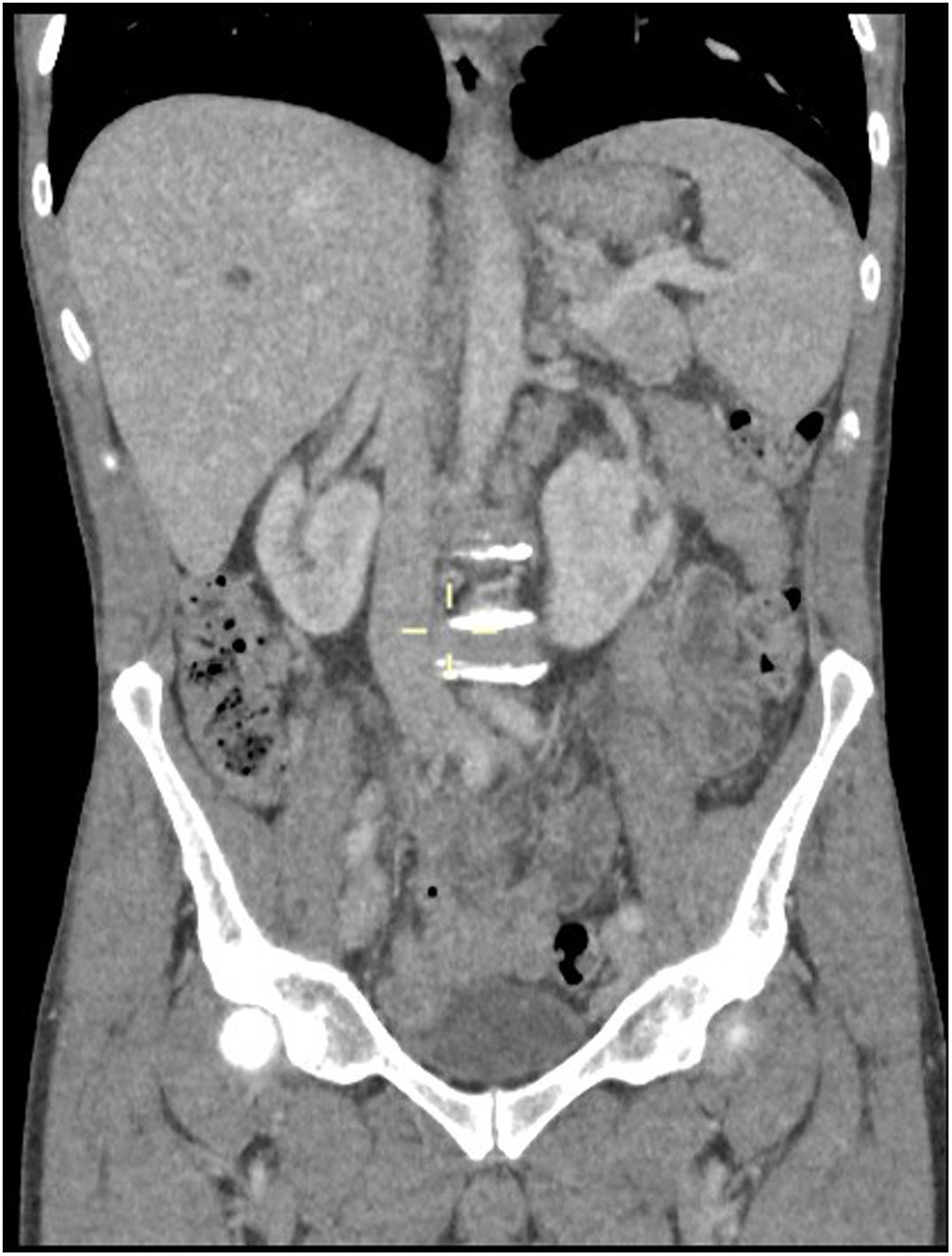
A whole-body CT scan showed generalized lymphadenopathy and moderate splenomegaly consistent with lymphoma (Figure 2). He had an excision biopsy of axillary lymph node that showed features of Angioimmunoblastic T-cell Lymphoma. A bone marrow biopsy showed element of T-cell marrow infiltration. A staging PET scan confirmed widespread lymphadenopathy with some tiny FDG positive cutaneous nodes on trunk (consistent with skin lesions) hence a stage IV-B.

CT chest, abdomen and pelvis; shows multiple enlarged lymph nodes likely pathological consistent with axillary and mediastinal and to a slightly lesser extent hilar lymphadenopathy. Paratracheal and subcarinal nodes measures 15mm in short-axis diameter. Right hilar and periesophageal nodes measures 14mm in diameter, left hilar and periesophageal measures 12mm in short-axis. Few mildly enlarged common femoral/external iliac lymph nodes bilaterally. Measuring at the short axis 17mm with an oval shape. Multiple retroperitoneal para-aortic, paracaval into aortocaval lymph nodes noted, measuring at the short axis between 10 and 14mm. There is also loss of definition around the portal confluence due to multiple periportal lymph nodes. Spleen is enlarged in the short axis at 8cm.
Patient was later followed up by oncology team and was discussed in the multidisciplinary team (MDT), and a decision was made to start CHOEP chemotherapy regimen for induction followed by autologous stem cell transplantation. Patient, however, declined chemotherapy as expected response was 30–50% only and was started on Prednisolone 50mg for symptomatic relief (a non-chemotherapeutic option).
In December 2017, he was admitted via emergency department due to jaundice with normal stools and no infective symptoms. His Hb was 69g/dL with normal WBC and PLT. His LFTs showed ALP 133, Bil 39, ALT 18, ALB 31, LDH 273, Serum Haptoglobin<0.10 (NR: 0.5–2.4). Direct Coombs test was positive ((IgG+C3d)) with cold agglutinins of IgM type (1:1076). A diagnosis of mixed autoimmune haemolytic anaemia was made. He was given 4 units of blood, Prednisolone was continued at 50mg for 2 weeks until haemoglobin levels were greater than 10g/dL (Hb 112g/dL, Bil<15, LDH 149 and serum Haptoglobin 0.9). A repeat CT scan showed no further progression of lymphoma.
He was continued on slowly tapering dose of Prednisolone (by 2.5–5mg/month) during the next 6 months under careful monitoring of haemoglobin and reticulocyte counts. Associated treatment included bisphosphonates, vitamin D, folic acid, enoxaparin and calcium.
A repeat Staging CT scan after 6 months showed complete resolution of lymphadenopathy and the splenomegaly. After remission and the patient taking 5mg of prednisolone daily, steroids were stopped without any significant adverse events. One year later on follow-up, laboratory tests of the patient were normal and when this article is written the patient remains well in complete remission without corticosteroid therapy
AITL is an uncommon subtype of mature peripheral T-cell lymphoma. It represents only 1–2% of all non-Hodgkin Lymphomas6 and 15–20% of Peripheral T-cell Lymphoma.7 It is a primary T-cell disorder with dysregulation noted in B-cells and endothelial cells as well.8 It has strong association with Epstein-Barr virus, found in 80–90%.9 It normally presents with B-type symptoms and lymphadenopathy, but rash is not an uncommon symptom as is seen in about 20–50% of patients7 and follows usually the introduction of antimicrobials.6 The rash of AITL most commonly comprise a maculopapular eruption on the trunk and abdomen (similar to our patient), but purpura, plaques, papulovesicular lesions, nodules, and erythroderma have also been reported. Approximately forty-four percent of patients have a nonspecific maculopapular dermatitis, which precedes other clinical symptoms by at least several weeks.10,11 AITL should be included in the differential diagnosis of any maculopapular eruption of unknown aetiology that is accompanied by lymphadenopathy. Given the high prevalence of the rash, it can be postulated that it is a specific manifestation of this lymphoma12 though oft ignored, and can be misdiagnosed as a non-specific viral rash in the absence of overt lymphadenopathy as in our case.
Our patient had all the B-symptoms, lymphadenopathy and the rash. The rash persisted even after the clinical symptoms and lymphadenopathy had resolved which is unusual. The association with Epstein-Barr virus was also noted.
Development of autoimmune haemolytic anaemia with both warm and cold agglutinins was another uncommon feature noted in this case which is found in only 13% of cases.7
Many agents have been used to treat AITL, however none has proved consistently effective, corticosteroids have long been first-line agents for AITL.
The patient had a favourable clinical outcome, achieving clinical remission after several months of oral corticosteroid monotherapy.
Given non-specific symptoms, AITL can be misdiagnosed as a non-specific viral illness, arthritis or even EBV given the association. The concern remains that a suspicion of AITL will require a keener eye of the physician to avoid delay in the diagnosis.
Conflicts of interestThe authors declare no conflicts of interest.


