
Hemophilia is well known in males, but poorly recognized in hemophilia carriers, who may have a hemorrhagic tendency, and the symptoms may be frequent and severe. Few studies have been done evidencing this bleeding in female carriers of the hemophilia gene.
ObjectivesTo verify the prevalence of hemorrhagic symptoms in HC, compared to women in the general population.
Material and methodThe articles published between October 1996 and November 2016 were searched in the PubMed, Scielo, Lilacs, Web of Science, Scopus and Cochrane Central databases.
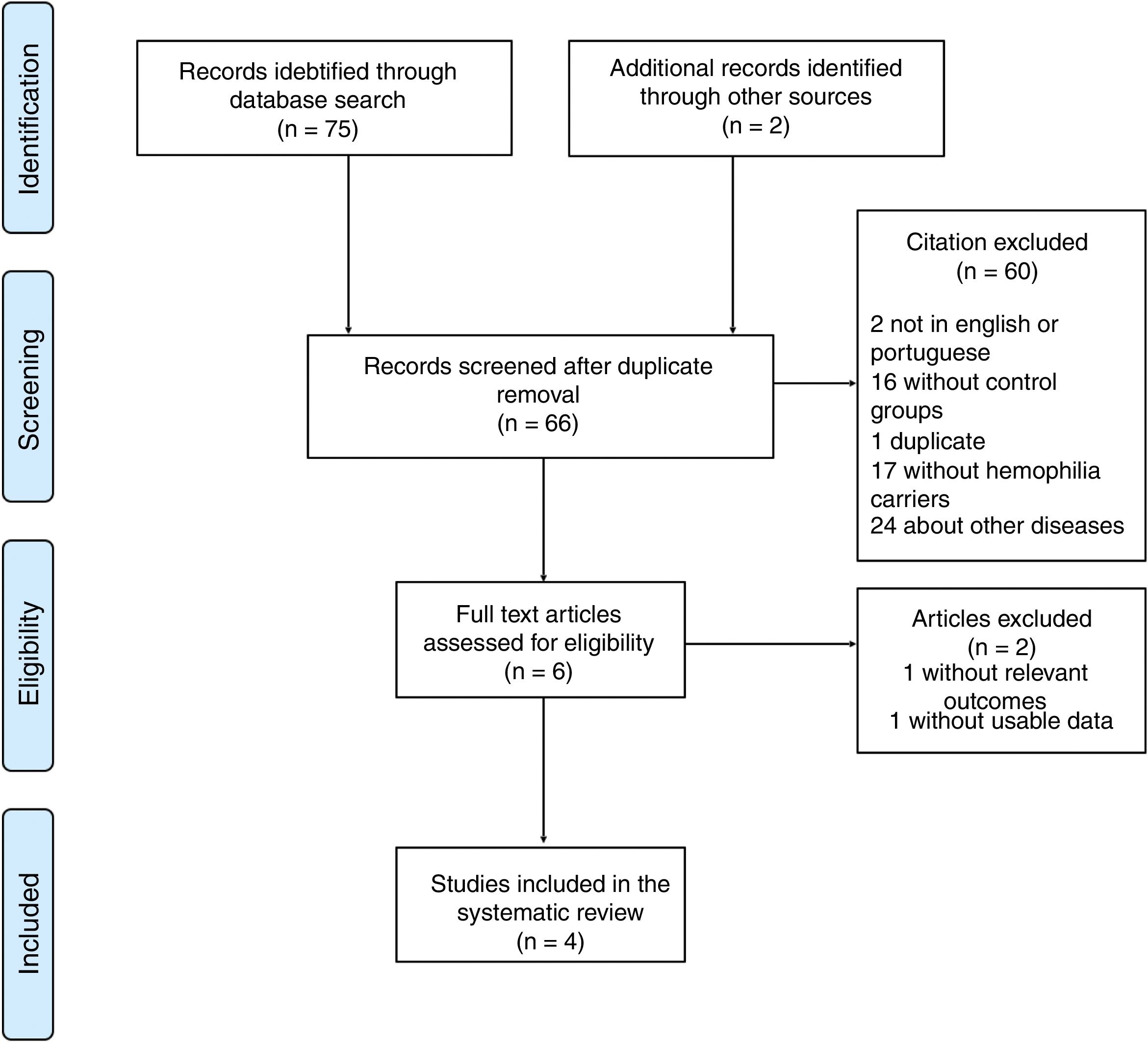
Results and discussionSeventy-five articles were found in electronic databases and 2 additional articles, through manual search in journal summaries and bibliographical references of other review articles. There is a limitation as to the number of studies that explore the association between the risk of hemorrhagic events and HC A or B. Among the few existing studies, there is a methodological difference, evidenced by control groups with distinct recruitments, divergent questionnaires and non-standardized concepts.
ConclusionThis review verified the existence of a higher prevalence of hemorrhagic symptoms in the HC in some outcomes, however, due to the limitations of the few studies found, there is still insufficient evidence to state that the HC has a greater hemorrhagic tendency in relation to the general population.
Hemophilia is an X - linked recessive disorder caused by a mutation in the F8 and F9 genes, resulting in deficient and/or defective production of factor VIII (FVIII) and factor IX (FIX) in Hemophilia A (HA) or Hemophilia B (HB), respectively. The coagulation is inadequate in a hemophilia patient, since these factors are deficient or the molecules are less functional as a consequence of mutations in the genes responsible for the production of these proteins. Both genes F8 and F9 are located in the distal portion of the long arm of the X chromosome at positions Xq28 and Xq27, respectively.1–6
Epidemiology shows an estimated hemophilia frequency of about 1/10,000 births. The prevalence of HA varies from 1/10,000 to 1/20,000 individuals and prevalence of HB is 1/30,000 to 1/50,000.2,5,8,9 In the 2015 Global Annual Survey of the World Federation of Hemophilia (WFH), 187,183 people worldwide were identified with hemophilia.10
The clinical picture is similar in both types of hemophilia, the differences being marked only by the amount of the deficient factor. The prevalent symptom is spontaneous or post-traumatic hemorrhage, musculoskeletal bleeding being the most common. The risk and severity of the bleeding are related to the deficient amount of the factor.7,11 Approximately two-thirds of patients have a family history of hemorrhagic symptoms. In addition, it has been established that 10 to 15% of hemophilia carriers (HCs) may have abnormal bleeding, which is not recognized by the patients and may, therefore, be overlooked by the physician and not be investigated.2,5,8,12,13 On the other hand, the HC frequently presents a propensity for hemorrhages and may therefore require treatment with clotting factor correction when subjected to hemorrhagic stress, a common occurrence for a hemophilic patient.14,15
The diagnosis of hemophilia is based on clinical history, family history, physical examination and laboratory tests, an exact diagnosis being essential for adequate treatment. Diagnostic confirmation occurs with the measurement of the deficient factor activity (FVIII or FIX). According to the degree of coagulant activity of these factors, hemophilia is characterized as mild (activity between 5 and 40%), moderate (between 1 and 5%) or severe (less than 1%).3,7,8
The HCs are currently recognized as a group that needs treatment, however, the physician awareness can be improved.There are few studies that investigated the association between the risk of bleeding events and the HC. In addition to this quantitative limitation, there is an important methodological difference among the studies included in this analysis, limiting the results of this systematic review. This suggests the need for further research on the subject.
Materials and methodsSearch strategyArticles published in English and Portuguese between October 1996 and November 2016 were searched in the electronic databases PubMed, Scielo, Lilacs, Web of Science, Scopus and Central Cochrane, using the search strategy P.I.C.O. (population, intervention/exposure, comparison, outcome) for each database. All citations were exported to the Mendeley Desktop (v. 1.16.1) for handling and duplicates were removed. The search strategy is described in Fig. 1.
Inclusion and exclusion criteria
Articles were eligible if they: (i) were written in English or Portuguese; (ii) had a comparison between the HC (A or B) and a control group; (iii) had symptomatic clinical outcomes, and; (iv) had proportions and averages for each group available.
Articles that did not meet these criteria were excluded from the analysis.
Selection of articlesThe initial selection of the articles was performed by ABH, AKCS and JSR, supervised and reviewed by MRFR. All the articles selected were read by the three authors (ABH, AKCS and JSR) to determine if they fit the inclusion criteria and the agreement between them (kappa=0.825). The discrepancies were resolved by consensus and mediated by MRFR.
Data acquisitionAfter the removal of duplicates, the data was extracted with Microsoft Office Excel worksheets. The authors that were contacted and did not provide the requested data were excluded from the data collection.
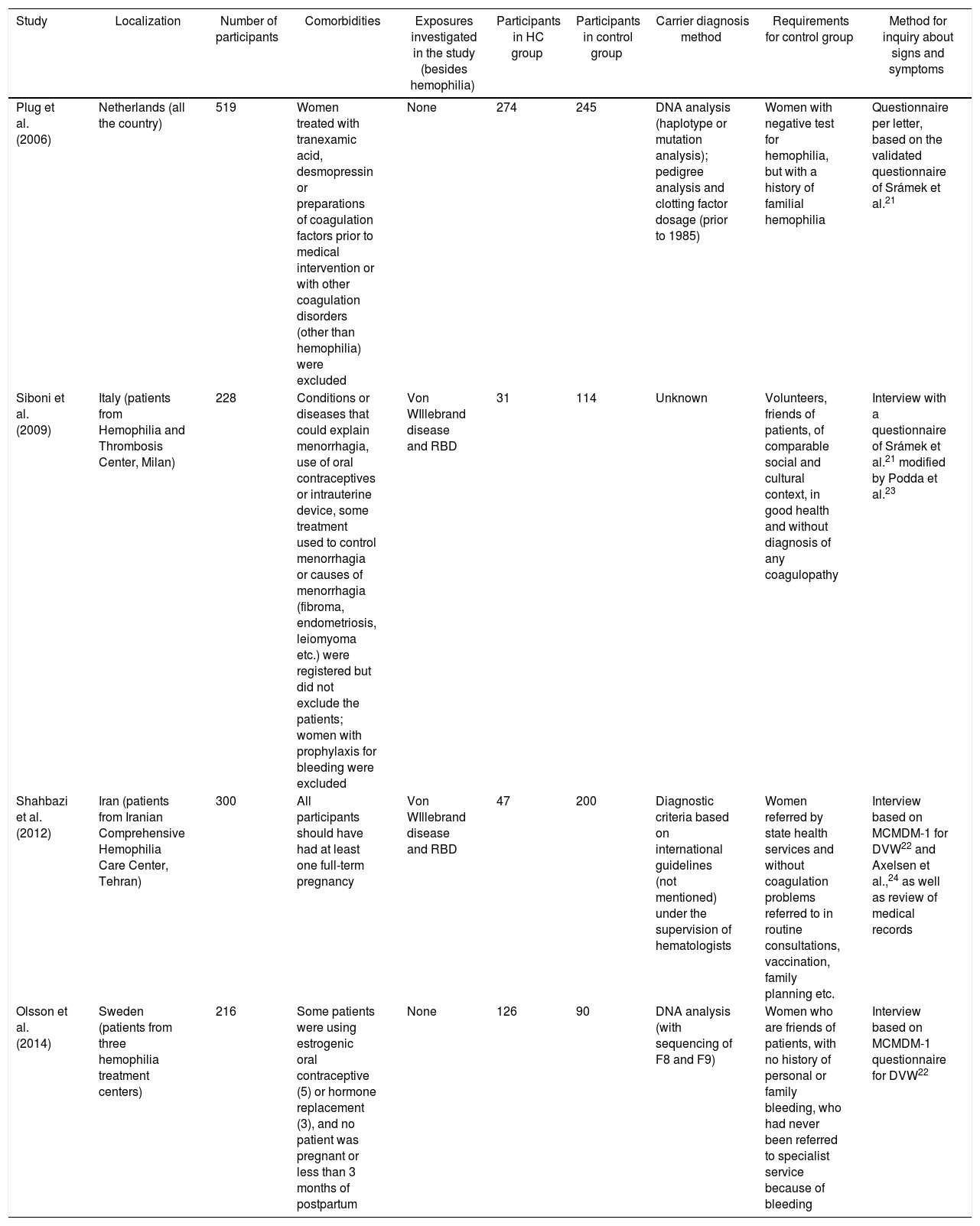
ParticipantsThe review sample consisted of HC A or B participants from four studies: Plug et al., 14 Siboni et al., 16 Shahbazi et al.17 and Olsson et al.18 The form of carrier recruitment was different among them: Plug et al. called all the women who were tested for HA and HB status at three reference centers for hemophilia in the Netherlands prior to 2001, considering as carriers those women diagnosed by DNA analysis (mutation or haplotype analysis) or factor coagulation measurement (prior to 1985), together with the pedigree analysis; Siboni et al. recruited positive HC patients at Angelo Bianchi Bonomi Hemophilia and Thrombosis Center (Milan, Italy); Shahbazi et al. recruited pregnant women from the Iranian Comprehensive Hemophilia Care Center with a positive diagnosis for hemophilia carrier; Olsson et al. analyzed HC A or B with the mutation confirmed by DNA analysis, recruited from three hemophilia treatment centers throughout Sweden.
The control group sample characteristics are in Table 1. The study of Paroskie et al.19 had participants, outcomes and a control group, but the data was incomplete; this manuscript was excluded because we could not obtain the necessary information for the analysis after contacting the authors.
Characteristics of studies selected for review.
| Study | Localization | Number of participants | Comorbidities | Exposures investigated in the study (besides hemophilia) | Participants in HC group | Participants in control group | Carrier diagnosis method | Requirements for control group | Method for inquiry about signs and symptoms |
|---|---|---|---|---|---|---|---|---|---|
| Plug et al. (2006) | Netherlands (all the country) | 519 | Women treated with tranexamic acid, desmopressin or preparations of coagulation factors prior to medical intervention or with other coagulation disorders (other than hemophilia) were excluded | None | 274 | 245 | DNA analysis (haplotype or mutation analysis); pedigree analysis and clotting factor dosage (prior to 1985) | Women with negative test for hemophilia, but with a history of familial hemophilia | Questionnaire per letter, based on the validated questionnaire of Srámek et al.21 |
| Siboni et al. (2009) | Italy (patients from Hemophilia and Thrombosis Center, Milan) | 228 | Conditions or diseases that could explain menorrhagia, use of oral contraceptives or intrauterine device, some treatment used to control menorrhagia or causes of menorrhagia (fibroma, endometriosis, leiomyoma etc.) were registered but did not exclude the patients; women with prophylaxis for bleeding were excluded | Von WIllebrand disease and RBD | 31 | 114 | Unknown | Volunteers, friends of patients, of comparable social and cultural context, in good health and without diagnosis of any coagulopathy | Interview with a questionnaire of Srámek et al.21 modified by Podda et al.23 |
| Shahbazi et al. (2012) | Iran (patients from Iranian Comprehensive Hemophilia Care Center, Tehran) | 300 | All participants should have had at least one full-term pregnancy | Von WIllebrand disease and RBD | 47 | 200 | Diagnostic criteria based on international guidelines (not mentioned) under the supervision of hematologists | Women referred by state health services and without coagulation problems referred to in routine consultations, vaccination, family planning etc. | Interview based on MCMDM-1 for DVW22 and Axelsen et al.,24 as well as review of medical records |
| Olsson et al. (2014) | Sweden (patients from three hemophilia treatment centers) | 216 | Some patients were using estrogenic oral contraceptive (5) or hormone replacement (3), and no patient was pregnant or less than 3 months of postpartum | None | 126 | 90 | DNA analysis (with sequencing of F8 and F9) | Women who are friends of patients, with no history of personal or family bleeding, who had never been referred to specialist service because of bleeding | Interview based on MCMDM-1 questionnaire for DVW22 |
There is a limitation of the number of studies that explore the association between the risk of hemorrhagic events and HC A or B. Among the few existing studies, there is a methodological difference, evidenced by control groups with distinct recruitments, divergent questionnaires and non-standardized concepts. One problem found in this review was the lack of general characteristics from the studies, because many of these had other interests such as: von Willebrand Disease (vWD), factor V Leiden, among others. Thus, questions such as age and ethnicity ended up being present in the sample, and not just the interest of the review (HC), which limited the comparison.
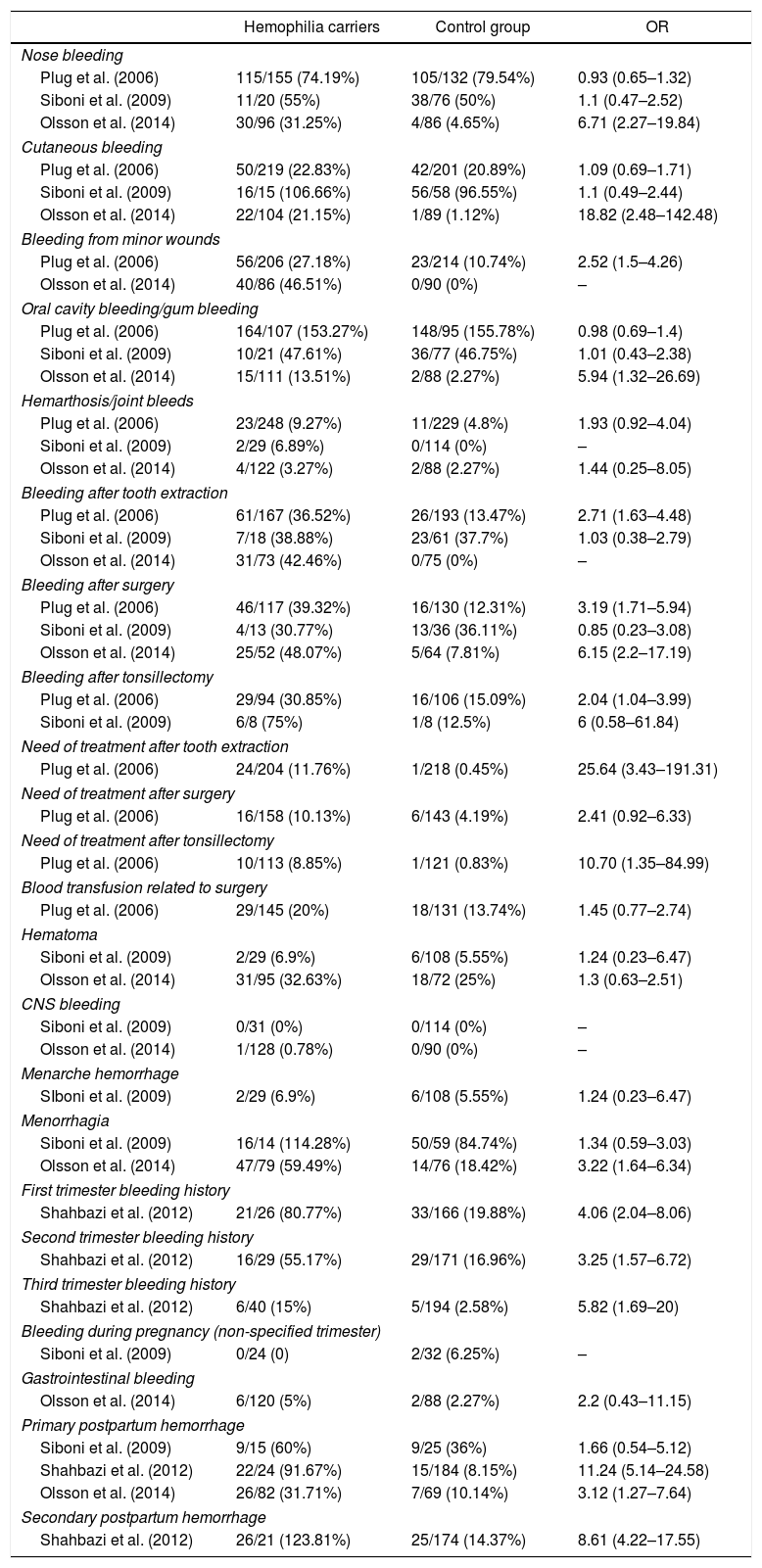
The odds and odds ratio (OR) for each outcome are in Table 2. The studies, although based on different questionnaires, had similar concepts, since they searched only in the absence or presence of the outcomes. However, Srámek et al.,21 Tosetto et al.22 and Podda et al.23 state that in clinical practice only the presence or absence of these outcomes is insufficient to predict the prognosis and the complete questionnaire is necessary. It is prudent to note that in the outcome “primary postpartum bleeding”, the concepts adopted by the three studies were expressively different, and only Shahbazi et al. used an objective definition, with temporal and volumetric delimitation, and therefore these works are theoretically not comparable in this outcome.
Odds and odds ratio, by outcome of interest, by hemophilia carriers and control groups, per study.
| Hemophilia carriers | Control group | OR | |
|---|---|---|---|
| Nose bleeding | |||
| Plug et al. (2006) | 115/155 (74.19%) | 105/132 (79.54%) | 0.93 (0.65–1.32) |
| Siboni et al. (2009) | 11/20 (55%) | 38/76 (50%) | 1.1 (0.47–2.52) |
| Olsson et al. (2014) | 30/96 (31.25%) | 4/86 (4.65%) | 6.71 (2.27–19.84) |
| Cutaneous bleeding | |||
| Plug et al. (2006) | 50/219 (22.83%) | 42/201 (20.89%) | 1.09 (0.69–1.71) |
| Siboni et al. (2009) | 16/15 (106.66%) | 56/58 (96.55%) | 1.1 (0.49–2.44) |
| Olsson et al. (2014) | 22/104 (21.15%) | 1/89 (1.12%) | 18.82 (2.48–142.48) |
| Bleeding from minor wounds | |||
| Plug et al. (2006) | 56/206 (27.18%) | 23/214 (10.74%) | 2.52 (1.5–4.26) |
| Olsson et al. (2014) | 40/86 (46.51%) | 0/90 (0%) | – |
| Oral cavity bleeding/gum bleeding | |||
| Plug et al. (2006) | 164/107 (153.27%) | 148/95 (155.78%) | 0.98 (0.69–1.4) |
| Siboni et al. (2009) | 10/21 (47.61%) | 36/77 (46.75%) | 1.01 (0.43–2.38) |
| Olsson et al. (2014) | 15/111 (13.51%) | 2/88 (2.27%) | 5.94 (1.32–26.69) |
| Hemarthosis/joint bleeds | |||
| Plug et al. (2006) | 23/248 (9.27%) | 11/229 (4.8%) | 1.93 (0.92–4.04) |
| Siboni et al. (2009) | 2/29 (6.89%) | 0/114 (0%) | – |
| Olsson et al. (2014) | 4/122 (3.27%) | 2/88 (2.27%) | 1.44 (0.25–8.05) |
| Bleeding after tooth extraction | |||
| Plug et al. (2006) | 61/167 (36.52%) | 26/193 (13.47%) | 2.71 (1.63–4.48) |
| Siboni et al. (2009) | 7/18 (38.88%) | 23/61 (37.7%) | 1.03 (0.38–2.79) |
| Olsson et al. (2014) | 31/73 (42.46%) | 0/75 (0%) | – |
| Bleeding after surgery | |||
| Plug et al. (2006) | 46/117 (39.32%) | 16/130 (12.31%) | 3.19 (1.71–5.94) |
| Siboni et al. (2009) | 4/13 (30.77%) | 13/36 (36.11%) | 0.85 (0.23–3.08) |
| Olsson et al. (2014) | 25/52 (48.07%) | 5/64 (7.81%) | 6.15 (2.2–17.19) |
| Bleeding after tonsillectomy | |||
| Plug et al. (2006) | 29/94 (30.85%) | 16/106 (15.09%) | 2.04 (1.04–3.99) |
| Siboni et al. (2009) | 6/8 (75%) | 1/8 (12.5%) | 6 (0.58–61.84) |
| Need of treatment after tooth extraction | |||
| Plug et al. (2006) | 24/204 (11.76%) | 1/218 (0.45%) | 25.64 (3.43–191.31) |
| Need of treatment after surgery | |||
| Plug et al. (2006) | 16/158 (10.13%) | 6/143 (4.19%) | 2.41 (0.92–6.33) |
| Need of treatment after tonsillectomy | |||
| Plug et al. (2006) | 10/113 (8.85%) | 1/121 (0.83%) | 10.70 (1.35–84.99) |
| Blood transfusion related to surgery | |||
| Plug et al. (2006) | 29/145 (20%) | 18/131 (13.74%) | 1.45 (0.77–2.74) |
| Hematoma | |||
| Siboni et al. (2009) | 2/29 (6.9%) | 6/108 (5.55%) | 1.24 (0.23–6.47) |
| Olsson et al. (2014) | 31/95 (32.63%) | 18/72 (25%) | 1.3 (0.63–2.51) |
| CNS bleeding | |||
| Siboni et al. (2009) | 0/31 (0%) | 0/114 (0%) | – |
| Olsson et al. (2014) | 1/128 (0.78%) | 0/90 (0%) | – |
| Menarche hemorrhage | |||
| SIboni et al. (2009) | 2/29 (6.9%) | 6/108 (5.55%) | 1.24 (0.23–6.47) |
| Menorrhagia | |||
| Siboni et al. (2009) | 16/14 (114.28%) | 50/59 (84.74%) | 1.34 (0.59–3.03) |
| Olsson et al. (2014) | 47/79 (59.49%) | 14/76 (18.42%) | 3.22 (1.64–6.34) |
| First trimester bleeding history | |||
| Shahbazi et al. (2012) | 21/26 (80.77%) | 33/166 (19.88%) | 4.06 (2.04–8.06) |
| Second trimester bleeding history | |||
| Shahbazi et al. (2012) | 16/29 (55.17%) | 29/171 (16.96%) | 3.25 (1.57–6.72) |
| Third trimester bleeding history | |||
| Shahbazi et al. (2012) | 6/40 (15%) | 5/194 (2.58%) | 5.82 (1.69–20) |
| Bleeding during pregnancy (non-specified trimester) | |||
| Siboni et al. (2009) | 0/24 (0) | 2/32 (6.25%) | – |
| Gastrointestinal bleeding | |||
| Olsson et al. (2014) | 6/120 (5%) | 2/88 (2.27%) | 2.2 (0.43–11.15) |
| Primary postpartum hemorrhage | |||
| Siboni et al. (2009) | 9/15 (60%) | 9/25 (36%) | 1.66 (0.54–5.12) |
| Shahbazi et al. (2012) | 22/24 (91.67%) | 15/184 (8.15%) | 11.24 (5.14–24.58) |
| Olsson et al. (2014) | 26/82 (31.71%) | 7/69 (10.14%) | 3.12 (1.27–7.64) |
| Secondary postpartum hemorrhage | |||
| Shahbazi et al. (2012) | 26/21 (123.81%) | 25/174 (14.37%) | 8.61 (4.22–17.55) |
The second edition of the Guidelines for the Management of Hemophilia, published by the WFH in 2012 on the conduct of the HC, considers that most hemophilia carriers are asymptomatic, using the levels of coagulation factors as indicators of symptomatic outcomes and, consequently, treatment.7 However, the monitoring of the HC with levels of coagulation factors is not accurate in predicting symptomatic outcomes with normal or subnormal clotting levels, being a helpful tool for the HC with low levels.18 Therefore, it is necessary to carefully monitor the carriers, so that the physician guides their behavior from a global assessment, which includes signs and symptoms, not just laboratory monitoring.
Siboni et al. excluded women who received clotting factor correction before an intervention, so this may have resulted in an underestimation of the bleeding tendency.
The control groups were not constructed in a standardized manner to represent the general population, since the recruitment tried to compare cases and controls only by general characteristics, such as age, profession, socioeconomic level, and so on. By not considering the familial character of the disease, an HC could have a greater fear of bleeding due to the very conviviality with hemophilic relatives, which would overestimate the symptoms in the group, or possibly underestimate the bleeding tendency, as more people have prolonged bleeding, which could be considered as normal.
Each of the study results was based on a different questionnaire, seeking only the presence or absence of symptomatic outcomes, with non-standardization of the concepts used in the evaluation of these outcomes and lack of objective criteria.
ConclusionThis review verified the existence of a higher prevalence of hemorrhagic symptoms in the HC in some outcomes, however, due to the inherent limitations of the few studies found, there is still insufficient evidence to state that the HC has a greater hemorrhagic tendency in relation to the general population, thus, it remains as a theoretical risk.
In the obstetric outcomes, the existing literature shows a higher prevalence of hemorrhagic symptoms in the pre- and postnatal period. Thus, clinical evaluation and follow-up of the pregnant HC are recommended, considering other risk factors for bleeding, even without consistent evidence.
The surgical outcomes were conflicting in previous studies. Pre- and postoperative care should be taken, but the importance of the uniqueness in the clinical approach for the HC is emphasized, as there is still insufficient evidence to universalize behaviors in this group.
Bleeding scales, such as those presented in the papers included in the review, are useful tools to verify the bleeding tendency, however, none of them have been validated for the HC. Thus, further studies are needed for the development of these tools. It is also necessary to adopt single concepts, objectives and representative control groups in future studies.
Although there is a great deal of information on bleeding in men with hemophilia, only a few studies have focused on bleeding in the HC, and this is sometimes an unrecognized issue. These findings highlight the importance of physicians and patients being aware of the complications that may occur after procedures that represent a great hemostatic challenge for the HC.
Conflicts of interestThe authors declare no conflicts of interest.
The authors stated that they had no interests which might be perceived as a conflict or bias.


