
Sickle cell disease is the most common monogenic disorder in humans and is a major public health concern in sub-Saharan Africa. In Benin, the prevalence of sickle cell disease is estimated to be 4.8%. Our study aimed to describe the prevalence of hemoglobin abnormalities in an apparently healthy Benin population.
MethodsOne thousand four hundred and eighty-three men and women, apparently in good health after medical screening, were tested for hemoglobin abnormalities by hemoglobin electrophoresis and the Emmel test. Subjects who were found to have homozygous or double heterozygous hemoglobin abnormalities, were re-sampled and a confirmation hemogram and hemoglobin electrophoresis test by capillary electrophoresis was performed.
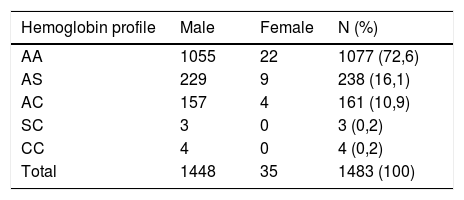
ResultsOur study population was predominantly male (97.7%) with an average age of 21.3 years. 1390 subjects reported that they did not know their hemoglobin electrophoresis status. Hemoglobin electrophoresis profiles found were as follows: 1077 (72.6%) AA (normal), 238 (16.1%) AS, 161 (10.9%) AC, 3 (0.2%) SC, 4 (0.2%) CC and 0 (0%) SS. The 406 subjects with abnormal hemoglobin had balanced somatic growth, with general physical examination results showing no abnormalities. In the seven subjects with major sickle cell syndrome or hemoglobinosis (SC and CC), their values of various hemogram parameters were normal apart from the discreet presence of microcytic anemia.
ConclusionOur study highlights the need for increased routine testing of hemoglobin abnormalities and newborn screening for sickle cell disease in order to enhance early disease detection, prevention and comprehensive care.
Sickle cell disease is the most common genetic disorder of hemoglobin in the world. Each year between 300,000 and 400,000 children are born with major sickle cell disease, particularly in sub-Saharan Africa, where it is a great public health problem.1,2
Biologically, sickle cell disease is marked by abnormal red blood cells caused by mutations in hemoglobin subunit beta genes. Under normal circumstances, a person inherits a copy of hemoglobin subunit beta genes (hemoglobin A) from each parent. However, if a person has sickle cell trait or disease, they inherit one or two copies of hemoglobin subunit beta gene that have a mutation respectively, which produces abnormal hemoglobin beta-globin such as hemoglobin S or C. Hemoglobin SS (homozygous) and hemoglobin SC (heterozygous) are amongst the most prevalent major sickle cell diseases.3 In Benin, the estimated prevalence of sickle cell trait (hemoglobin S) is 22.3% and hemoglobin C is 10.21% and approximately 4% of the population in Benin is affected by hemoglobin SS homozygosity and hemoglobin SC double heterozygosity.4
In Benin, systematic screening for sickle cell disease is not a common practice, as there are few laboratories capable of conducting a correct diagnosis. Diagnosis is normally made by clinical symptoms; however, carriers of sickle cell trait often are asymptomatic.
The objective of our study was to determine the prevalence of hemoglobin abnormalities in young Beninese people who are asymptomatic. The rationale was to underscore the importance of focused and enhanced prevention efforts for sickle cell disease and systematic newborn screening.
MethodsThis is a retrospective descriptive study conducted in 2016. The study population consisted of subjects of both sexes who applied for recruitment into the military. Recruitment required assessment of medical fitness of the candidates in 2 stages. First, a brief physical examination was conducted by a nurse involving a running endurance test. Secondly, selected recruits received a medical examination performed by a doctor, along with clinical testing. For the candidates who received the medical examination, we performed a hemoglobin electrophoresis blood test (to detect abnormal hemoglobin) and an Emmel test, also known as a sickling test (to screen for Hemoglobin S by detecting changes in the shape of red blood cells).
Subjects who were found to have a homozygous or double heterozygous hemoglobin abnormality were re-sampled and had a hemogram and hemoglobin electrophoresis test performed for confirmation.
All clinical tests were performed at the laboratory of the Hôpital d’Instruction des Armées in Cotonou. A blood sample was drawn for each subject by venipuncture. Subjects were advised to have an empty stomach for sample collection. Blood was collected in tubes with EDTA (ethylenediaminetetraacetic acid) and the following tests were performed: hemoglobin electrophoresis using cellulose acetate, (pH 8.6),5 hemogram using the Mindray BC-3000 Plus as per manufacturer instructions and the Emmel test using blood from a finger stick and mixing the blood with sodium metabisulfite.5 Samples with a homozygous or double heterozygous anomaly were confirmed by capillary electrophoresis using Minicap, Sebia, as per manufacturer instructions.
Finally, for each subject, we collected demographic (age, sex, background, consanguinity) and clinical information (weight, height, general condition).
Statistical analysisStatistical analyses (number, frequency, average) were carried out using Excel and Epi-info software version 4.2.
Ethical considerationsTests were carried out according to the local regulations in place with the agreement of the military authorities concerned. Prior to the initiation of any study procedures, background information was provided to all study subjects, from whom informed consent was also obtained. Patient confidentiality and data privacy were ensured throughout the study by deidentifying all subject data and using deidentified subject codes during the testing process. The study complied with good clinical practice protocols and with the ethics rules stated in the Declaration of Helsinki.
ResultsOne thousand four hundred and eighty-three (1483) samples were analyzed by hemoglobin electrophoresis. 97.7% (n=1448) of the subjects were male. The average age of the study population was 21.3 years with an age range of 18–25 years. All participants reported being in good apparent health and reported no past surgical history or known hemoglobinopathy. 1390 participants had never received a hemoglobin electrophoresis test. Table 1 summarizes the different electrophoresis profiles found. A homozygous SS genotype was not found in any of the subjects.
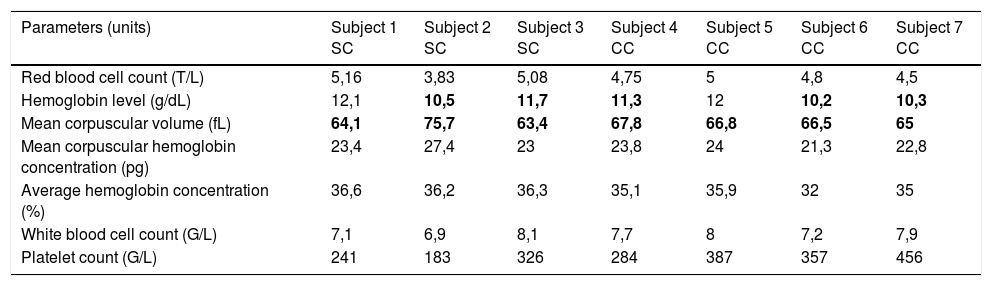
The 406 subjects with abnormal hemoglobin (AS, AC, SC, CC) had regular somatic growth and a normal physical examination. For the seven subjects with major sickle cell syndrome or hemoglobinosis (SC and CC), a hemogram was performed. The values of the various blood count parameters are shown in Table 2. A common abnormality found in the subjects with major sickle cell syndrome or hemoglobinosis was constant microcytosis anemia, sometimes associated with mild anemia.
Hematological characteristics of subjects with SC or CC hemoglobin abnormalities. Numbers that are in bold indicate values outside of the normal laboratory reference range.
| Parameters (units) | Subject 1 SC | Subject 2 SC | Subject 3 SC | Subject 4 CC | Subject 5 CC | Subject 6 CC | Subject 7 CC |
|---|---|---|---|---|---|---|---|
| Red blood cell count (T/L) | 5,16 | 3,83 | 5,08 | 4,75 | 5 | 4,8 | 4,5 |
| Hemoglobin level (g/dL) | 12,1 | 10,5 | 11,7 | 11,3 | 12 | 10,2 | 10,3 |
| Mean corpuscular volume (fL) | 64,1 | 75,7 | 63,4 | 67,8 | 66,8 | 66,5 | 65 |
| Mean corpuscular hemoglobin concentration (pg) | 23,4 | 27,4 | 23 | 23,8 | 24 | 21,3 | 22,8 |
| Average hemoglobin concentration (%) | 36,6 | 36,2 | 36,3 | 35,1 | 35,9 | 32 | 35 |
| White blood cell count (G/L) | 7,1 | 6,9 | 8,1 | 7,7 | 8 | 7,2 | 7,9 |
| Platelet count (G/L) | 241 | 183 | 326 | 284 | 387 | 357 | 456 |
Laboratory reference ranges for an adult male subject: Red blood cell count (4-6T/L); hemoglobin level (12–16g/dL); mean corpuscular volume (89–90fL); mean corpuscular hemoglobin concentration (25–32pg); average hemoglobin concentration (30–36%); white blood cell count (3–8G/L), platelet count (150–400G/L).
We studied the distribution of hemoglobin abnormalities in young people from Benin, aged 18 to 25 years old, who were in apparent good health. This study was conducted in order to better inform health education initiatives for the prevention of hemoglobin disorders, including for couples who are not yet married.
The study population consisted of 1483 subjects, predominantly male (97.7%) with an average age of 21.3 years. The subjects were military personnel recruited for military service who were aware that they had to be in good physical condition to serve in the military. The sample size and population of this study was defined by the number of study recruits who received physical assessments.
Amongst our study population, the prevalence of S and C sickle cell trait carriers was 16.1% and 10.9%, respectively. These rates are similar to those previously reported in Benin by Latoundji et al. (22.3% and 10.1%, respectively)4 and more recently by Noudamadjo et al. in infants in 2013 (14.64% and 14.20%).6 In west Africa, similar rates are also reported in Togo by Segbena et al. (16.4% S sickle cell and 15.8% C sickle cell)7 and Kafando et al. in Burkina Faso (17% S sickle cell and 21% C sickle cell).8 It should be noted that high prevalence of hemoglobin C in Burkina Faso is explained by the fact that it is the home from which hemoglobin C originates.9
Although SC heterozygosity is an integral part of major sickle cell syndromes, it presents as less severe than SS homozygosity.10,11 However, the natural evolution of SC heterozygous is marked by the occurrence of often irreversible complications whose pathophysiology involves existing hyper-viscosity and hypercoagulability of blood.12,13 The most common of these complications are: aseptic osteonecrosis of the femoral and humeral heads, ocular damage (retinal detachment, retinal hemorrhage). The tendency of clinical symptoms to diminish and the discreteness of hematologic abnormalities often results in delayed diagnosis, with affected individuals failing to be diagnosed until adulthood.13 Data in literature indicates that height-weight development of SC heterozygous is not significantly different compared to the general reference population, but there is a consistent growth delay in people with Hb SS type.11 Biologically, however, constant microcytic anemia is present in Hb SS or SC type. Literature indicates that microcytic anemia in type SC is constant, though not very severe (10–12g/dL). Ngo Sack et al in their series of reports on SC patients, reported a prevalence of 47.4% microcytic anemia.10 The severity of microcytosis in our study also suggests the concurrent existence of iron deficiency. Iron deficiency remains a major public health problem in developing countries such as Benin.14 Systematic screening for sickle cell and thalassemia must also be considered. Therefore, in the presence of microcytosis and hypochromia, iron assessment should be undertaken before considering iron therapy.14
A prevalence of 0.2% Hb CC type was found in our study, similar to that found by Noudamadjo et al. in Benin.6 Hemoglobin CC appears more common in North Africa with a reported prevalence of 1.6% and 8%, respectively, by Mseddi et al. in Tunisia15 and Ouzzif et al. in Morocco.16 The discovery of the genotypic profile CC often occurs late in one’s life, if at all, due to its asymptomatic nature. According to Ngouadjeu et al., hemoglobinosis CC is a tolerable, often asymptomatic condition with moderate hypochromic microcytic anemia.17 The symptomatology when it is present is atypical or attenuated and is manifested by pallor, sub-jaundice, bone pain and splenomegaly. In the face of manifestations of sickle-cell disease, the diagnosis of CC hemoglobinosis must be thoroughly clinically examined.17
SS homozygous sickle cell disease is distinguished by the severity of its clinical presentation with acute and chronic complications. These recurring manifestations appear early in infancy, impact the development of weight and height and can lead to organic failures and disabling functional sequelae.18 Our study population included healthy adults who were recruited into the military, which explains the absence of hemoglobin SS.
Given the complex disease manifestation of hemoglobin abnormalities, it is therefore important to remind medical staff to examine quantitative and qualitative abnormalities of blood counts and to undertake the necessary follow-up investigations for the rapid detection and diagnosis of a large number of pathologies, including composite heterozygous sickle cell disease (hemoglobin SC). In Benin, improved clinical care for hematological pathologies is being a focus of the National Agency for Blood Transfusion (ANTS) of Benin, whose 4th edition in February 2018 was devoted to improved blood count monitoring.19 Our study shows that a significant proportion of apparently healthy young adults do not know their hemoglobin profile. More than 25% of subjects are carriers of sickle cell trait, carrying a hemoglobin abnormality. It is known that early detection of hemoglobin profile, associated with parental education, and comprehensive care, markedly reduces morbidity and mortality from sickle cell disease.20 Newborn screening program complement and enhance clinical services. Our results reinforce the newborn screening as the most important and efficient hemoglobinopathies tracking policy. The newborn screening program is implemented in Benin since 1993.21 It is demontred significantly decrease sickle cell disease related morbidity and mortality rates, as already shown in developed countries.21
These clinical considerations highlight the need for an increased focus on hemoglobin abnormalities by medical personnel in order to provide patients with clear information so that they may seek services for early detection of hemoglobinopathies, leading to enhanced prevention for sickle cell disease. The need for enhanced screening and prevention of hemoglobin abnormalities, which are complex, and detrimental though asymptomatic, is underscored in our study in which the three subjects who had SC abnormalities presented with a normal phenotype but may never been diagnosed with major sickle cell syndrome due to lack of symptoms. As such, we propose enhanced efforts to better inform population to adhere to newborn screening program in order to improve prevention and management of hemoglobin disorders.
Contribution of the authorsA.Z. (Faculté des Sciences de la Santé, Centre National Hospitalier Universitaire-HKM, Hôpital d’Instruction des Armées de Cotonou-CHU) is the initiator of the study and wrote the manuscript; T.A-B., L.Z. et L.A. (Faculté des Sciences de la Santé, Centre National Hospitalier Universitaire-HKM) read, commented and contributed to the improvement of the manuscript.
Conflicts of interestThe authors declare no conflicts of interest.
We would like to thank Remi Charlebois, Michele Merkel and Rachel Crane (Global Scientific Solutions for Health) for manuscript review, proofreading, and translation from French to English.


